
Cклеродермия у детей | Интернет-издание «Новости медицины и фармации»
Склеродермия (СД) — это полисиндромное заболевание, проявляющееся прогрессивным фиброзом кожи, внутренних органов и сосудистой патологией.
Патогенез
Патогенез СД включает следующие основные моменты:
1. Нарушение функции фибробластов: ускорение биосинтеза коллагена, формирование аномальных коллагеновых волокон.
2. Поражение мелких сосудов: облитерация мелких артерий, артериол, капилляров, нарушение микроциркуляции, нарушение строения и функции пораженной ткани.
3. Аутоиммунные сдвиги: образование аутоантител к коллагену, ядрам клеток, эндотелию сосудов, мышцам.
Классификация и номенклатура
СД у детей традиционно классифицировалась как ювенильная локализованная склеродермия (JLS) и ювенильный системный склероз (JSS). JLS в дальнейшем классифицировалась как очаговая (morphea), локализованная или генерализованная и линейная СД, включая поражения в виде следа после удара саблей «en coup de sabre» по лбу и Parry-Romberg гемифациальную атрофию.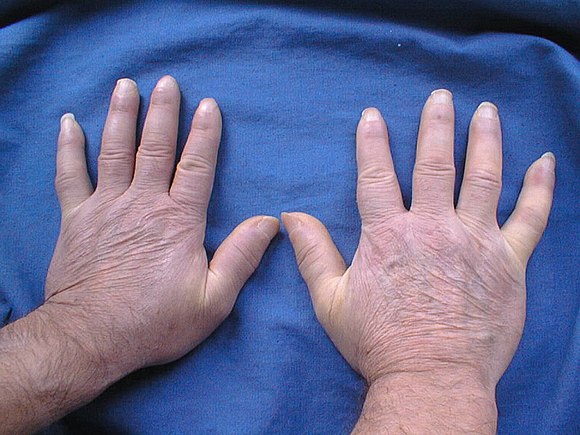
Ограниченная склеродермия может быть бляшечной и полосовидной. В ней выделяют очаговую форму (1–5 очагов), диссеминированную (6–30 очагов), распространенную — сливную (бляшки и полосы поражают лицо, конечности и значительную часть туловища) и генерализованную (без поражения внутренних органов) (Никитина, 1980).
Эпидемиология
СД — редкое заболевание с частотой 0,05 на 100 000 детей. Средний возраст больных составляет 8 лет, 90 % педиатрических больных имеют диффузный характер поражения.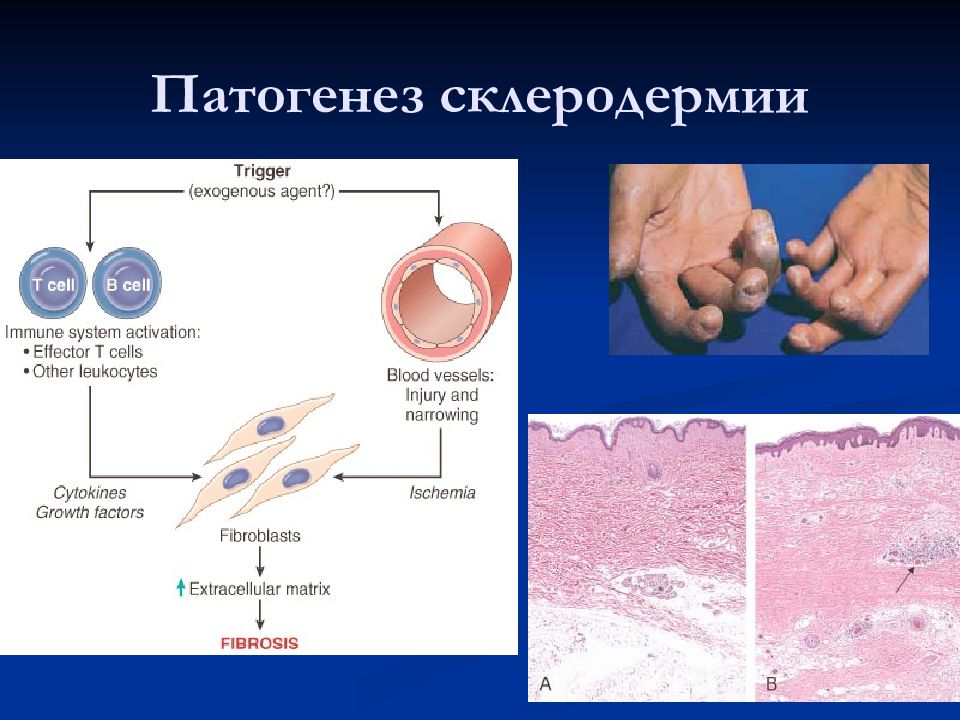
Некоторые авторы выделяют Боррелиа-ассоциированную форму бляшечной очаговой склеродермии, характеризующуюся началом болезни в раннем возрасте, инфицированием B. Burgedorfi и выраженным аутоиммунным феноменом, который проявляется высоким титром антинуклеарных антител. Болезнь имеет тяжелое течение и требует лечения как инфекции, так и кожного воспаления [6].
Поднимается вопрос о врожденной склеродермии. Как заявил д-р. Lawrence Schachner на ежегодной педиатрической конференции Университета Майами, врожденная локализованная склеродермия — редкий диагноз, который, возможно, просматривается у маленьких детей. Он сообщил, что при мультинациональном анализе историй болезни 750 детей с ювенильной линейной СД установлено, что у 6 больных (0,8 %) впервые клинические и серологические признаки склеродермии были обнаружены сразу после рождения.
Клинические особенности склеродермии у детей
Ювенильная локализованная склеродермия обычно считается болезнью, ограниченной кожей и подкожной клетчаткой. Группа авторов (F. Zulian et al.) [10] и рабочая группа по ювенильной склеродермии Европейского педиатрического ревматологического общества (PRES) изучали преобладание и клинические симптомы внекожных проявлений у большой группы детей с ювенильной локализованной склеродермией. Под наблюдением находилось 750 больных. У 168 из них (22,4 %) были выявлены 193 внекожных поражения, в том числе суставные (47,2 %), неврологические (17,1 %), сосудистые (9,3 %), глазные (8,3 %), гастроинтестинальные (6,2 %), респираторные (2,6 %), кардиальные (1 %) и ренальные (1 %).
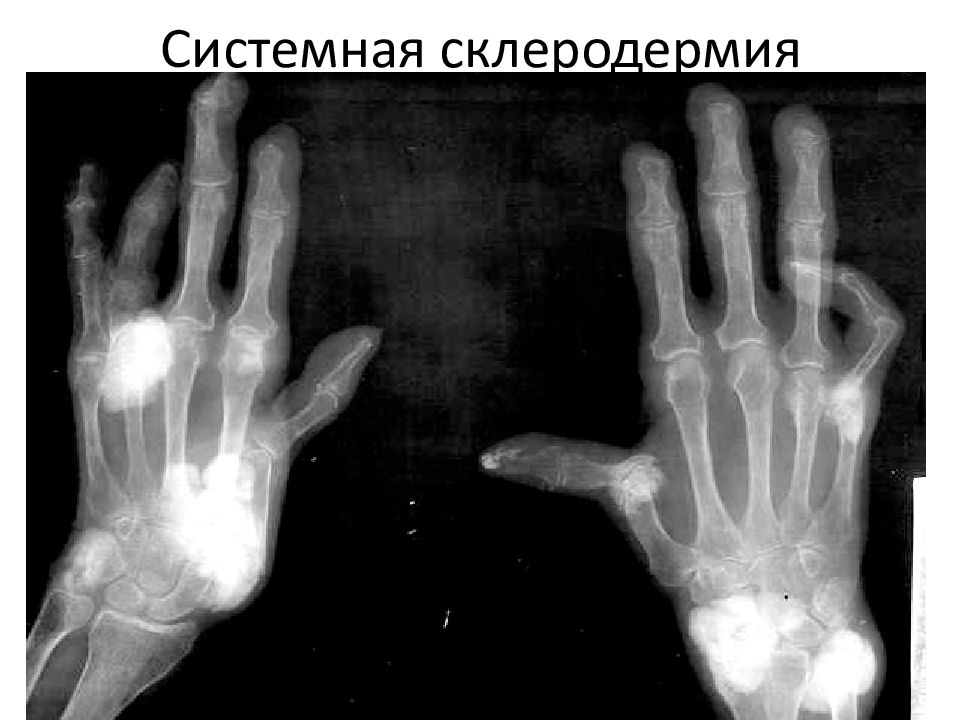
По сравнению со взрослыми дети с атакой ювенильного системного склероза чаще имеют смешанные (overlap) синдромы с полимиозитом (РМ) — дерматомиозитом, большую частоту вовлечения скелетной мускулатуры, наличие анти-РМ-СД и анти-U1-RNP антител, фатальной кардиальной патологии и более высокую выживаемость [8].
Мультифакториальный анализ факторов, влияющих на выживаемость при СД, основанный на изучении 134 историй болезни из 40 центров, дал следующие результаты: 16 больных умерли, 4 — в течение 1 года после установления диагноза и 10 — в течение 5 лет. В качестве значимых предикторов смертности отмечены фиброз на рентгенограмме грудной клетки, повышение уровня креатинина и перикардит. Все больные с фатальным исходом были поражены диффузной формой болезни и у большинства из них отмечались быстрое прогрессирование и ранние признаки вовлечения в процесс внутренних органов.
Лечение склеродермии у детей
Патогенетические звенья различных форм СД определены только частично, но главный дефект при СД — это аномальные коллагеновые депозиты, ведущие в конечном результате к фиброзу кожи, а также внутренних органов — сердца, легких — при JSS. Следовательно, терапевтические мероприятия для лечения СД можно разделить на 3 главные группы: антифибротические, противовоспалительные средства и вазодилататоры. Для локализованных форм болезни противовоспалительные средства, аналоги витамина D и УФО — в стадии изучения. Тем не менее нечастая СД у детей плюс факт, что болезнь очень часто дает спонтанные ремиссии, делает рандомизированные контролируемые исследования очень трудными. По этой причине большинство данных о терапевтических программах при этой болезни у детей были получены из результатов исследований у взрослых.
В стадии изучения находятся такие новые методы лечения этого сложного заболевания, как трансплантация аутологичных стволовых клеток и цитокинкорригирующая терапия [7].
С целью изучения особенностей течения СД на современном этапе и эффективности терапии нами было проведено специальное исследование, в ходе которого изучены особенности болезни 3 детей, находившихся на лечении по поводу СД в соматическом отделении ГДКБ № 16 г. Харькова. Из них 2 девочки и 1 мальчик в возрасте 9–14 лет.
Установлено, что первыми ранними признаками болезни у всех детей являлись очаговые поражения кожи и подкожных тканей, локализующиеся на одной стороне (по данным реовазографии). Патологический процесс сопровождался поражением желудочно-кишечного тракта (в виде хронического гастродуоденита, дуоденогастрального рефлюкса, дискинезии желчевыводящих путей у 1 ребенка), поражением почек (в виде дисметаболической нефропатии у всех детей), поражением сердца (в виде диспластической кардиопатии, вегетососудистой дисфункции у 2 детей), поражением щитовидной железы (в виде диффузного зоба, эутиреоза у 1 ребенка), изменением функциональной адгезивности тромбоцитов (в виде тромбоцитопатии) у всех исследуемых детей.
Выводы
С помощью рентгенологических исследований могут выявляться нарушения моторики пищевода и тонкой кишки. Функциональное исследование легких, ЭКГ, рентгенография позволяют выявить поражение сердечной и дыхательной систем. При поражении почек отмечаются изменения в анализах мочи и нарушение функции почек.
При поражении почек отмечаются изменения в анализах мочи и нарушение функции почек.
Эффективность лечения в значительной мере определяется не только ранним началом терапии, но и ее непрерывностью, что важно учитывать при ведении ребенка.
Bibliography1. Bernstein R.M., Pereira R.S., Holden A.J., Black C.M., Howard A., Ansell B.M. Autoantibodies in childhood scleroderma // Ann. Rheum. Dis. — 1985. — 44 (8). — 503-6.
2. Christen-Zaech S., Hakim M.D., Afsar F.S., Paller A.S. Pediatric morphea (localized scleroderma): review of 136 patients. — Depatment of Dermatology, Northwestern University Feinberg School of Medicine, Chicago, Illinois 60611-2997, USA. — 2008. — 59 (3). — 385-96.
3. De Macedo P.A., Shinjo S.K., Goldenstein-Schainberg C. Juvenile scleroderma //Acta Reumatol Port. — 2008. — 33 (3). — 289-97.
4. Foeldvari I. Current development in pediatric systemic sclerosis // Curr. Rheumatol Rep. — 2009. — 11 (2). — 97-102.
5. Martini G. , Vittadello F., Kasapcopul O. et al. Factors affecting survival in juvenile systemic sclerosis // Rheumatology (Oxford). — 2009. — 48 (2). — 119-22.
, Vittadello F., Kasapcopul O. et al. Factors affecting survival in juvenile systemic sclerosis // Rheumatology (Oxford). — 2009. — 48 (2). — 119-22.
6. Prinz J.K., Kutasi Z., Weisenseel P., Poto L., Battyani Z., Ruzicka T. Borrelia — associated early — onset morphea: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? Results of a cohort analysis and presentation of three cases // J. am Acad. Dermatol. — 2009. — 60 (2). — 248-55.
7. Rosenkranz M.E., Agic L.M., Efthimiou P., Lehman T.J. Systemic and localized scleroderma in children: current and future treatment options // Paediatr. Drugs. — 2006. — 8 (4). — 270.
8. Scalapino K., Arkachaisr i T., Lucas M., Fertig N. Childhood onset systemic sclerosis: classification, clinical and serologic features, and survival in comparison with adult onset disease // J. Rheumatol. — 2006. — 33 (5). — 1004-13.
9. Woo P. Theoretical and practical basis for early aggressive therapy in paediatric autoimmune disorders // Curr. Opin. Rheumatol. — 2009. — 29.
Opin. Rheumatol. — 2009. — 29.
10. Zulian F., Martini G. Childhood systemic sclerosis // Curr. Opin. Rheumatol. — 2007. — 19 (6). — 592-7.
11. Zulian F., Vallongo C., Woo P., Russo R. et al. Localised scleroderma in childhood is not just a skin disease // Arthritis Rheum. — 2005. — 52 (9). — 2873-81.
Системная склеродермия — диагностика и лечение в СПб
ЛЕЧЕНИЕ СИСТЕМНОЙ СКЛЕРОДЕРМИИ ДОСТУПНО В ФИЛИАЛАХ:
Лечение системной склеродермии в Приморском районе
Адрес: г. Санкт-Петербург, Приморский район, ул. Репищева, 13
Лечение системной склеродермии в Петроградском районе
Адрес: г. Санкт-Петербург, Петроградский район, ул. Ленина, 5
Системная склеродермия — редкое хроническое ревматическое заболевание, в результате которого происходит уплотнение кожи, соединительной ткани, а так же возможно поражение кровеносных сосудов и внутренних органов. Чаще возникает у женщин в возрасте от 30 до 50 лет. Кроме того, болезни подвержены дети. Системная склеродермия отличается непредсказуемостью течения (от состояния ремиссии на протяжении многих лет до быстрого поражения всего организма). Только ранняя диагностика и правильное лечение склеродермии даст возможность контролировать заболевание и сохранить нормальную жизнь.
Чаще возникает у женщин в возрасте от 30 до 50 лет. Кроме того, болезни подвержены дети. Системная склеродермия отличается непредсказуемостью течения (от состояния ремиссии на протяжении многих лет до быстрого поражения всего организма). Только ранняя диагностика и правильное лечение склеродермии даст возможность контролировать заболевание и сохранить нормальную жизнь.
Причины системной склеродермии
Уплотнение ткани возникает в результате чрезмерного синтеза и накопления коллагена. Природа системной склеродермии до конца не изучена, однако предполагается, что причиной являются аутоиммунные процессы, когда организм начинает воспринимать собственные клетки как чужеродные.
Среди причин выделяют:
- генетическую предрасположенность,
- нарушения синтеза коллагена
- травмы
- инфекции
Симптомы системной склеродермии
Выделяют несколько типов склеродермии:
- Очаговая – ограниченные уплотнения на конечностях и лице.
 Очаговая склеродермия может вовлекать мышцы и кости, внутренние органы не затрагиваются.
Очаговая склеродермия может вовлекать мышцы и кости, внутренние органы не затрагиваются. - Системная — более агрессивный вариант болезни, при котором характерно повреждение кровеносных сосудов с сопутствующими признаками (синдром Рейно):
 Очаговая склеродермия может вовлекать мышцы и кости, внутренние органы не затрагиваются.
Очаговая склеродермия может вовлекать мышцы и кости, внутренние органы не затрагиваются.- Бледность пальцев, кончика носа на холоде
- Ощущения покалывания или онемения
На поздних стадиях кожа становится натянутой и блестящей, возникает проблема с сжатием пальцев в кулак
- Уплотнение кожи
- Потеря волосяного покрова в местах уплотнения
- Депигментация
При поражении сердечной ткани (нарушение сердцебиения, перебои в сердце) может возникнуть перикардит, миокардит, инфаркт.
Нарушения в легких ведёт к фиброзу тканей (возникновение сильной отдышки и стойкого сухого кашля).
Наличие системной склеродермии может привести к осложнениям при беременности.
Диагностика
При подозрении на склеродермию следует немедленно обратиться к ревматологу или дерматологу. В этом случае необходимо комплексное обследование:
В этом случае необходимо комплексное обследование:
- лабораторные анализы (кровь и моча)
- ЭКГ
- Эхо-КГ
- рентген грудной клетки
- спирография
Лечение системной склеродермии
Лечение склеродермии зависит от степени тяжести заболевания. В периоды обострения назначается медикаментозная терапия (противовоспалительная, сосудорасширяющая, иммуномодулирующая). Широко применяются физиотерапия, парафиновые аппликации, массаж, ЛФК.
Приём ведут врачи:
Выберите филиал“Династия” на Новочеркасском пр-те, Красногвардейский район“Династия” на Ленина, Петроградский район“Династия” на Репищева, Приморский район“Династия” во ВсеволожскеВыездная служба
Стоимость лечения системной склеродермии:
| Наименование услуг | Цена в рублях | |
| Санкт-Петербург | Всеволожск | |
| Первичный прием ревматолога 1 ступени | 1850 | — |
| Повторный прием ревматолога 1 ступени | 1650 | — |
| Первичный прием ревматолога, ведущего специалиста | 2500 | 2000 |
| Повторный прием ревматолога, ведущего специалиста | 2300 | 1800 |
| МАНИПУЛЯЦИИ | ||
| Капилляроскопия | 1500 | — |
ЗАПИСЬ НА ЛЕЧЕНИЕ СИСТЕМНОЙ СКЛЕРОДЕРМИИ
Ваша заявка отправлена
Менеджер свяжется с вами для уточнения деталей
Мы ценим ваше обращение в наш медицинский центр «Династия»
Ограниченная склеродермия — ФГБНУ НИИР им.
 В.А. Насоновой
В.А. НасоновойОграниченная склеродермия
Что такое Ограниченная склеродермия?
Ограниченная склеродермия обычно ограничивается локальным поражением кожи без системности процесса, т.е. у пациента отсутствует синдром Рейно и поражение внутренних органов.
Различают две основные формы заболевания:
-
Очаговую склеродермия (бляшечная форма; генерализованная форма)
-
Линейная склеродермия (полосовидная; типа «удара саблей»; фасциальная гемиатрофия Ромберга)
Линейная склеродермия развивается, как правило, у детей и подростков, а очаговая склеродермия может выявиться в любом возрасте, иногда генерализованное поражение кожи туловища начинается в преклимактерическом периоде.
Симптомы
В зависимости от характера и распространения кожного поражения очаговая склеродермия подразделяется на бляшечную форму и генерализованную форму. Чаще встречается бляшечная форма склеродермии, характеризующаяся наличием одного и более очагов различной локализации (туловище, конечности) чаще белого цвета с лиловым ободком в активный период заболевания, кожа в этом месте может уплотняться и быть спаяна с подлежащими тканями или наоборот атрофична по типу «папирусной бумаги», иногда имеются участки гиперпигментации. Генерализованная форма характеризуется практически тотальным поражением кожи туловища и конечностей.
Чаще встречается бляшечная форма склеродермии, характеризующаяся наличием одного и более очагов различной локализации (туловище, конечности) чаще белого цвета с лиловым ободком в активный период заболевания, кожа в этом месте может уплотняться и быть спаяна с подлежащими тканями или наоборот атрофична по типу «папирусной бумаги», иногда имеются участки гиперпигментации. Генерализованная форма характеризуется практически тотальным поражением кожи туловища и конечностей.
Линейная склеродермия делится на полосовидную форму и склеродермию по типу «удара саблей». Полосовидная форма локализуется чаще на конечностях, нередко при этой форме поражаются подлежащие коже ткани: фасции, мышцы, околосуставные ткани; возможно развитие контрактур суставов. При фасциальной гемиатрофии Ромберга отмечается односторонняя атрофия подкожной клетчатки на лице, которая может сопровождаться неврологической сиптоматикой. При склеродермии типа «удара саблей» поражается кожа лица и волосистой части головы, могут вовлекаться в процесс кости черепа, появится глазная и неврологическая симптоматика.
Лабораторные сдвиги чаще отсутствуют, но иногда выявляются антинуклеарные антитела.
Специалисты, занимающиеся диагностикой и лечением в институте: сотрудники лаборатории микроциркуляции и воспаления
Запишитесь на приём к специалисту:
Системная склеродермия — эффективное лечение в Москве
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначить только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Статья проверена врачом-ревматологом Филатовой Е.Е., носит общий информационный характер, не заменяет консультацию специалиста.
Для рекомендаций по диагностике и лечению необходима консультация врача.
Врачи Клинического госпиталя на Яузе проводят всестороннюю комплексную диагностику системной склеродермии и разрабатывают оптимальную программу лечения для каждого пациента (при помощи противовоспалительных, антифиброзных, сосудистых средств, препаратов, нормализующих иммунитет, гравитационной хирургии крови).
Системная склеродермия — это прогрессирующее заболевание, в результате которого поражается соединительная ткань организма. Начинается более активная выработка белка-коллагена, соединительная ткань уплотняется, наблюдаются признаки фиброза. Женщины болеют системной склеродермией примерно в 7 раз чаще, чем мужчины. В большинстве случаев заболевание встречается у пациентов в возрасте 30-50 лет.
Симптомы системной склеродермии
Являясь патологией соединительной ткани, системная склеродермия имеет множество различных симптомов, так как заболевание может поражать практически любой орган (соединительная ткань присутствует во всех органах).
Однако можно выделить наиболее часто встречающиеся признаки болезни:
- спазм сосудов пальцев — часто предшествует началу заболевания
- отек кожи и ее уплотнение
- атрофия участков кожи
- образование очагов склероза в коже
- уплотнение стенок сосудов, повышение вероятности образования тромбов
- значительное снижение веса (на 10 кг и более)
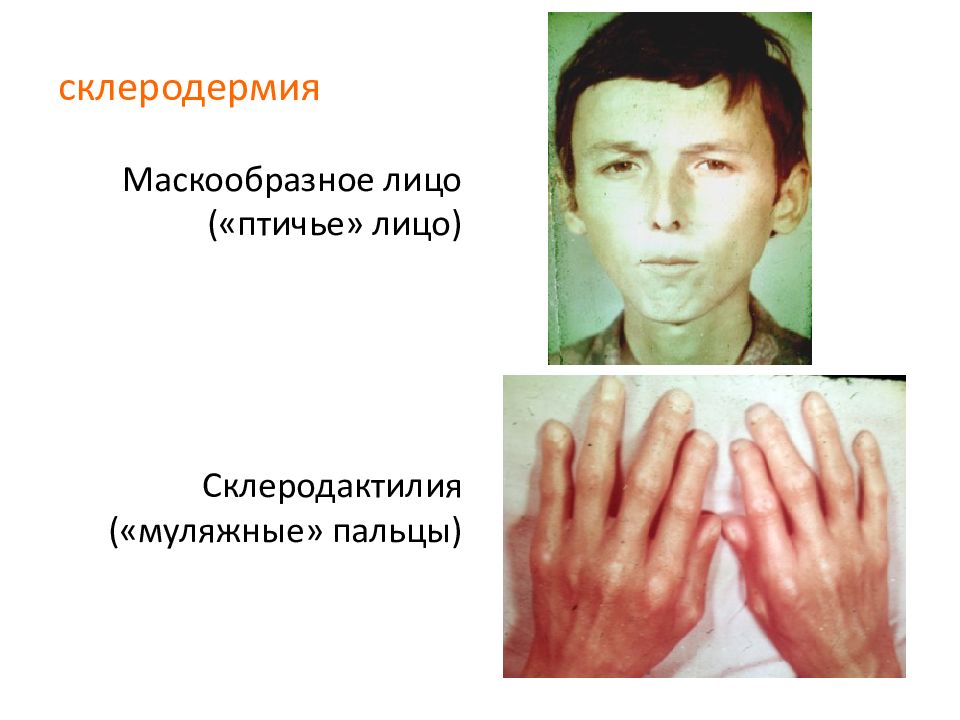
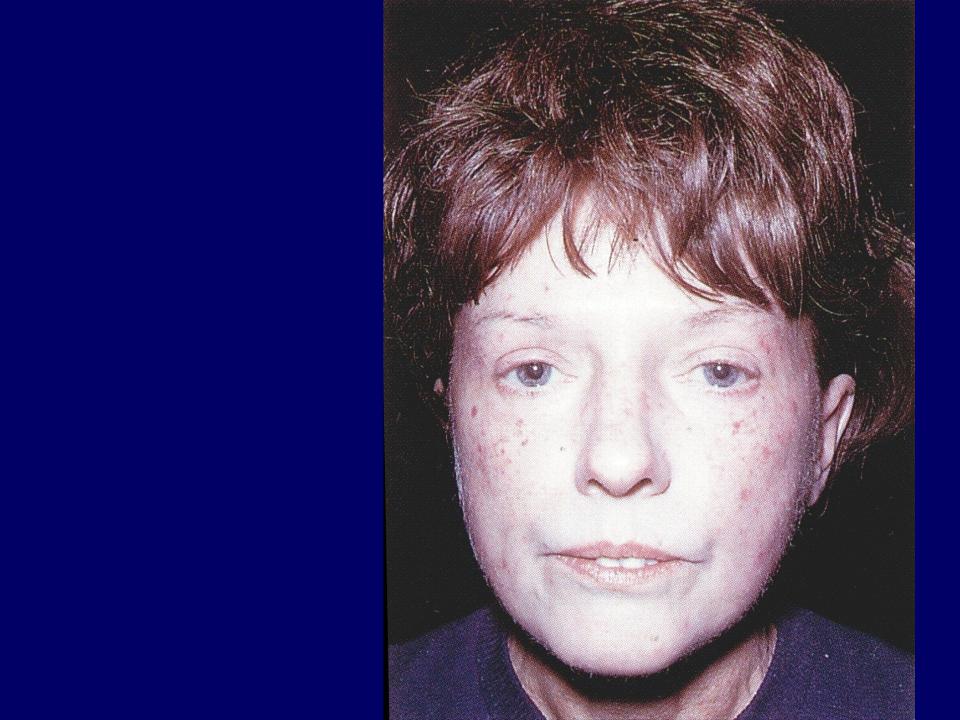
У больных системной склеродермией отмечаются характерные изменения внешности: нос заостряется («клювовидный нос»), вокруг рта образуются складки, кожа на кистях рук натягивается.
Также при системной склеродермии отмечаются нарушения со стороны сердечно-сосудистой системы, почек, развивается гипертония. У некоторых пациентов заболевание затрагивает слизистые оболочки, у них начинается конъюнктивит, стоматит и т.д.
Причины и патогенез системной склеродермии
Как отмечают специалисты, точные причины системной склеродермии пока не известны.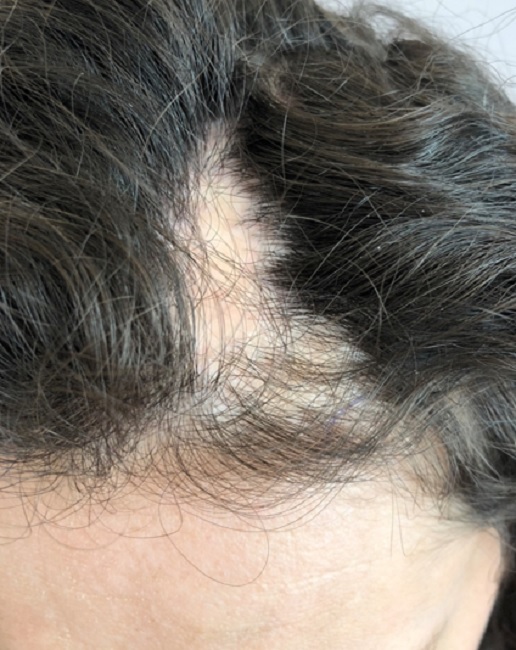 Не исключено, что заболевание имеет вирусную природу, так как в пораженных тканях при анализе обнаруживаются вирусоподобные частицы, а в крови — противовирусные антитела. Однако доказательств этой теории пока нет.
Не исключено, что заболевание имеет вирусную природу, так как в пораженных тканях при анализе обнаруживаются вирусоподобные частицы, а в крови — противовирусные антитела. Однако доказательств этой теории пока нет.
К факторам, которые способствуют развитию системной склеродермии, относятся:
- генетическая предрасположенность
- снижение иммунитета
- эндокринные нарушения
- склонность к аллергии
- переохлаждение
- травма
- стресс
- условия труда (вибрация, контакт с нежелательными веществами, например, силиконовой пылью)
Диагностика и лечение системной склеродермии в Клиническом госпитале на Яузе
Системная склеродермия диагностируется по совокупности внешних проявлений, а также на основании лабораторных и инструментальных исследований:
- анализ крови, в том числе с составлением иммунограммы
- анализ мочи
- биопсия кожи
- исследование внутрисуставной жидкости
- ЭКГ
- УЗИ внутренних органов
Лечение системной склеродермии заключается в приеме противовоспалительных, антифиброзных, сосудистых препаратов, а также препаратов, нормализующих иммунитет.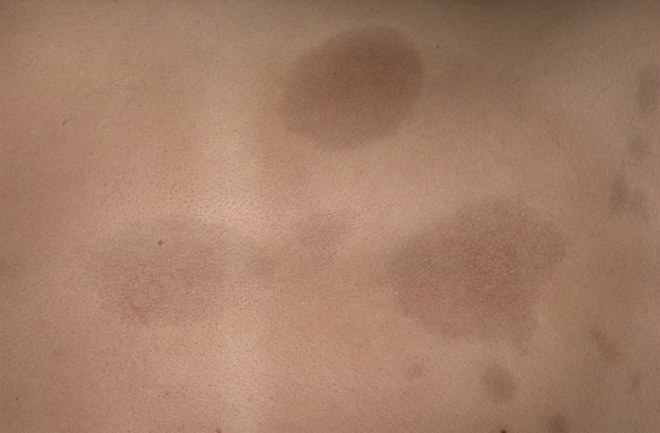 Высокую эффективность при лечении системной склеродермии показали методы экстракорпоральной гемокоррекции, которые позволяют удалить из крови аутоантитела, способствующие развитию заболевания.
Высокую эффективность при лечении системной склеродермии показали методы экстракорпоральной гемокоррекции, которые позволяют удалить из крови аутоантитела, способствующие развитию заболевания.
В рамках лечения пациентам показаны гимнастика, массаж и местные виды терапии.
Лечение системной склеродермии методами гемокоррекции
Методы экстракорпоральной гемокоррекци позволяют удалять из крови патологические компоненты, а также снижать дозы медикаментов при повышении их эффективности.
Специалисты Клинического госпиталя на Яузе применяют:
- сочетание криоафереза и инкубации клеточной массы;
- каскадную фильтрацию плазмы;
- лимфоцитаферез;
- фотоферез.
Стоимость услуг
Цены на услуги Вы можете посмотреть в прайсе или уточнить по телефону, указанному на сайте.
ВОПРОСЫ КЛАССИФИКАЦИИ, КЛИНИЧЕСКАЯ КАРТИНА И БАЗИСНАЯ ТЕРАПИЯ ЮВЕНИЛЬНОЙ СКЛЕРОДЕРМИИ
218
этому среди детских ревматологов прочно укоренилось
мнение о возможности и необходимости влиять на тяже-
ло протекающие ограниченные формы болезни средст-
вами БТ. По данным опроса детских ревматологов Се-
По данным опроса детских ревматологов Се-
верной Америки (2010), для лечения ЮОСД чаще назна-
чают МТ в дозе 10–12 мг/м2в неделю совместно с ГК
или в виде монотерапии, реже – ММФ, в торпидных
случаях – сочетание МТ и ММФ [24]. Известно, что
в указанных дозах МТ обладает антипролиферативной
активностью, угнетает иммунное воспаление, достаточ-
но хорошо переносится детьми. Многочисленными экс-
периментальными и клиническими исследованиями
[25–27] доказано, что при парентеральном способе вве-
дения МТ обладает большей эффективностью, по срав-
нению с пероральным его приемом, за счет высокой
биодоступности, которая при пероральном приеме пре-
парата зависит, помимо прочего, от характера пищи.
Последнее обстоятельство вынуждает рекомендовать
прием МТ внутрь за 1 ч до еды либо на ночь, что часто
вызывает усиление свойственных МТ желудочно-ки-
шечных расстройств. Использование МТ в предзапол-
ненных шприцах для подкожного введения (методжект)
во многом помогает повысить эффективность лечения
и снизить частоту неблагоприятных лекарственных ре-
акций. Длительность терапии МТ, по данным литерату-
Длительность терапии МТ, по данным литерату-
ры, составляет 1–3 года. При инициации лечения МТ
может назначаться совместно с ГК. Нет единства во
мнении о дозе ГК, разные специалисты используют до-
зы от 0,5 до 1 мг/кг в сутки, чаще на 8–12 нед, с полной
отменой через 1–6–12 мес. Имеется описание назначе-
ния ГК в стартовой дозе 2 мг/кг в сутки на 2 нед, с после-
дующим медленным снижением и отменой к 12 мес [28].
Доказанная эффективность МТ в лечении ЮОСД,
по нашему мнению, не должна исключать ПА из арсена-
ла средств БТ. Наш опыт применения монотерапии ПА
при очаговой бляшечной ЮОСД в фазе индуративного
отека свидетельствует о способности препарата ликви-
дировать индурацию и поверхностный фиброз в очаге,
при этом эффект лечения развивается медленно, к 12-му
месяцу терапии. Общая длительность монотерапии ПА
составляет не менее 3 лет [29]. Возможность успешного
лечения некоторых форм ЮОСД без использования ГК
и цитостатиков мы считаем принципиально важной.
При наличии противопоказаний к назначению либо не-
переносимости МТ у детей с распространенными бля-
шечными, линейными очагами, гемисклеродермией
комбинация ПА и ГК также проявила себя как достаточ-
но эффективная. По-видимому, специфика лечения
ЮСД в Российской Федерации, в условиях холодного
климата и высокой заболеваемости детей интеркуррент-
ными инфекциями, такова, что ограничивает примене-
ние цитостатиков, поэтому использование ПА сохраня-
ет свою актуальность. Некоторые детские ревматологи
за рубежом [30] в настоящее время тоже широко исполь-
зуют ПА.
Таким образом, вопрос формулировки четких реко-
мендаций к выбору варианта БТ при конкретной клиниче-
ской форме ЮОСД остается открытым. Ведется междуна-
родное исследование по стандартизации плана лечения
ЮОСД в первые 12 мес терапии, в соответствии с опубли-
кованным в 2012 г. [28] консенус-планом, предусматрива-
ющим использование ГК, МТ, ММФ.
Показанием для назначения препаратов БТ является
активный период ЮССД, а также ЮОСД с быстрым про-
грессированием кожного процесса, поражением мышц,
суставов, гемисклеродермия туловища и лица, особенно
в сочетании с эписиндромом и/или увеитом. Проводимые
в настоящее время в мире исследования по стандартизации
БТ ЮС, вероятно, позволят выявить оптимальный вари-
ант и длительность лечения для каждой формы болезни,
улучшить прогноз заболевания.
Прозрачность исследования
Исследование не имело спонсорской поддержки. Авторы
несут полную ответственность за предоставление оконча-
тельной версии рукописи в печать.
Декларация о финансовых и других взаимоотношениях
Все авторы принимали участие в разработке концеп-
ции статьи и в написании рукописи. Окончательная версия
рукописи была одобрена всеми авторами. Авторы не получали
гонорар за статью.
Педиатрическая ревматология
1. Никитина МН. Ограниченная склеродермия у детей.
Ограниченная склеродермия у детей.
Вопросы клиники, патогенеза и лечения. Автореф. дисс. …
д-ра мед.наук. Москва;1980 [Nikitina MN. Ogranichennaya
sklerodermiya u detei. Voprosy kliniki, patogeneza i lecheniya.
Avtoref. diss. … d-ra med.nauk [Limited scleroderma in children.
Questions clinic, pathogenesis and treatment: Dr. Diss. (Med.
Sci.)]. Moscow; 1980].
2. Zulian F. New developments in localized scleroderma. Curr Opin
Rheumatol. 2008;20(5):601–7. doi:
10.1097/BOR.0b013e328309a5eb
3. Pelkonen PM, Jalanco HJ, Lantto RK, et. al. Incidence of sys-
temic connective tissue disease in children: a nationwide prospec-
tive study in Finland. J Rheumatol. 1994;21(11):2143–6.
4. Herrick AL, Ennis H, Bhushan M, et al. Incidence of childhood
linear scleroderma and systemic sclerosis in the UK and Irelands.
Arthritis Care Res (Hoboken). 2010;62(2):213–8. doi:
10.1002/acr.20070
5. Atzeni F, Bardoni A, Cutolo M, et. al. Localized and systemic
al. Localized and systemic
forms of scleroderma in adults and children. Clin Exper Rheumatol.
2006:24(1 Suppl 40):36–45.
6. Foeldvari I. Juvenile systemic sclerosis. In: Varga J, Denton CP,
Wigley FM, editors. Scleroderma: from pathogenesis to compre-
hensive management. New York: Springer Science+Business
Media, LLC; 2012. 93 p.
7. Martini G, Foelvari I, Russo R, et al. Systemic sclerosis in child-
hood: clinical and immunologic features of 153 patients. Arthritis
Rheum. 2006;54(12):3971–8. doi: 10.1002/art.22207
8. Zulian F, Athreya B, Laxer R, et al. Juvenile localized scleroder-
ma: clinical and epidemiological features in 750 children. An
international study. J Rheumatol. 2006;45(5):614–20. doi:
10.1093/rheumatology/kei251
9. Cassidy TJ, Petty RE. Text book of Pediatric Rheumatology.
Toronto: Elsevier Saunders Сompany; 2002. 505 p.
10. Уварова НН. Клиническая картина и течение системной
склеродермии у детей. Автореф. дисс. … д-ра мед. наук.
Автореф. дисс. … д-ра мед. наук.
Москва; 1989 [Uvarova NN. Klinicheskaya kartina i techenie sis-
temnoi sklerodermii u detei. Avtoref. diss. … d-ra med. nauk [The
clinical picture and course of systemic sclerosis in children: Dr.
Diss. (Med. Sci.)]. Moscow; 1989].
11. Гусева НГ. Системная склеродермия
и псевдосклеродермические синдромы. Москва: Медицина;
1993. С.25. [Guseva NG. Sistemnaya sklerodermiya i psev-
ЛИТЕРАТУРА
Немецкие клинические рекомендации по диагностике и лечению локализованной склеродермии
Локализованная склеродермия представляет собой гетерогенную группу склерозирующих заболеваний кожи. В зависимости от подтипа, тяжести течения заболевания, локализации очага отмечается вовлечение в патологический процесс различных структур – жировой, мышечной ткани, суставов, костей. Данная статья является обновлением текущих рекомендаций German AWMF (Association of the Scientific Medical Societies in Germany). В рекомендациях рассматривается определение, эпидемиология, классификация, патогенез, лабораторная диагностика, гистологическая картина, клинические системы оценки заболевания, инструментальная диагностика локализованной склеродермии. Более того, в статье приводятся рекомендации по лечению пациентов в зависимости от клинического варианта заболевания. Рекомендации представлены в форме алгоритма. Фармакологические компании не оказывали финансовой поддержки во время разработки рекомендаций. Рекомендации являются актуальными до июля 2019г.
Более того, в статье приводятся рекомендации по лечению пациентов в зависимости от клинического варианта заболевания. Рекомендации представлены в форме алгоритма. Фармакологические компании не оказывали финансовой поддержки во время разработки рекомендаций. Рекомендации являются актуальными до июля 2019г.
Определение
Локализованная склеродермия, также известная как морфеа, объединяет гетерогенную группу склерозирующих заболеваний кожи. В зависимости от подтипа, тяжести течения заболевания, локализации очага отмечается вовлечение в патологический процесс различных структур – жировой, мышечной ткани, суставов, костей. При этом, не наблюдается поражения внутренних органов – сердца, легких, почек, ЖКТ, а также прогрессирования заболевания в системный склероз.
! Диагностика и лечение заболевания должны осуществляться при содействии группы квалифицированных специалистов – дерматолога, ревматолога, детского ревматолога и/или педиатра.
Эпидемиология и классификация
Частота встречаемости локализованной склеродермии, по данным литературы, составляет около 27 случаев на 1 млн человек. В исследовании, проведенном на территории Англии и Ирландии частота встречаемости заболевания составила 3,4 случаев на 1 млн детей. Локализованная склеродермия у женщин встречается в 2.6-6 раз чаще, чем у мужчин. Широкий спектр клинических проявлений заболевания привел к появлению большого количества различных классификаций. Авторы данных рекомендаций предложили классификацию, учитывающую тяжесть, распространенность и глубину процесса фиброзирования, выделив пять основных клинических вариантов: ограниченную, генерализованную, линейную, глубокую и смешанную склеродермию (табл 1). Преимуществом такой упрощенной классификации является взаимосвязь с тактикой лечения, представленной в данных рекомендациях.
В исследовании, проведенном на территории Англии и Ирландии частота встречаемости заболевания составила 3,4 случаев на 1 млн детей. Локализованная склеродермия у женщин встречается в 2.6-6 раз чаще, чем у мужчин. Широкий спектр клинических проявлений заболевания привел к появлению большого количества различных классификаций. Авторы данных рекомендаций предложили классификацию, учитывающую тяжесть, распространенность и глубину процесса фиброзирования, выделив пять основных клинических вариантов: ограниченную, генерализованную, линейную, глубокую и смешанную склеродермию (табл 1). Преимуществом такой упрощенной классификации является взаимосвязь с тактикой лечения, представленной в данных рекомендациях.
Классификация также в некоторой мере отражает особенности клинического течения различных форм заболевания. Например, у 50% пациентов с ограниченной локализованной склеродермией через 2,5 года от начала заболевания отмечается регресс клинических проявлений. Напротив, более длительное течение заболевания (около 5,5 лет) характерно для генерализованной, линейной и глубокой склеродермии. Однако, приведенные значения описывают лишь усредненные данные. Регресс вторичных изменений – гипер- и депигментации, контрактуры, атрофические изменения обычно незначителен и происходит крайне медленно. Как правило, частота встречаемости определенных подтипов зависит от возраста (например, линейная локализованная склеродермия чаще встречается в детском возрасте). Дети могут иметь несколько форм заболевания одновременно, например, линейная локализованная склеродермия в комбинации с ограниченной локализованной склеродермией. Наличие двух форм заболевания должно отражаться в диагнозе.
Однако, приведенные значения описывают лишь усредненные данные. Регресс вторичных изменений – гипер- и депигментации, контрактуры, атрофические изменения обычно незначителен и происходит крайне медленно. Как правило, частота встречаемости определенных подтипов зависит от возраста (например, линейная локализованная склеродермия чаще встречается в детском возрасте). Дети могут иметь несколько форм заболевания одновременно, например, линейная локализованная склеродермия в комбинации с ограниченной локализованной склеродермией. Наличие двух форм заболевания должно отражаться в диагнозе.
Ограниченная форма локализованной склеродермии
Наиболее распространенной формой локализованной склеродермии является бляшечная склеродермия (морфеа). Наиболее типичной локализацией является туловище, особенно складки под молочной железой, переход от паха в бедренную область. Очаги часто имеют овальную форму, на ранних стадиях заболевания могут казаться эритематозными. впоследствии они прогрессивно уплотняются в центре и приобретают беловатый либо кремовый оттенок.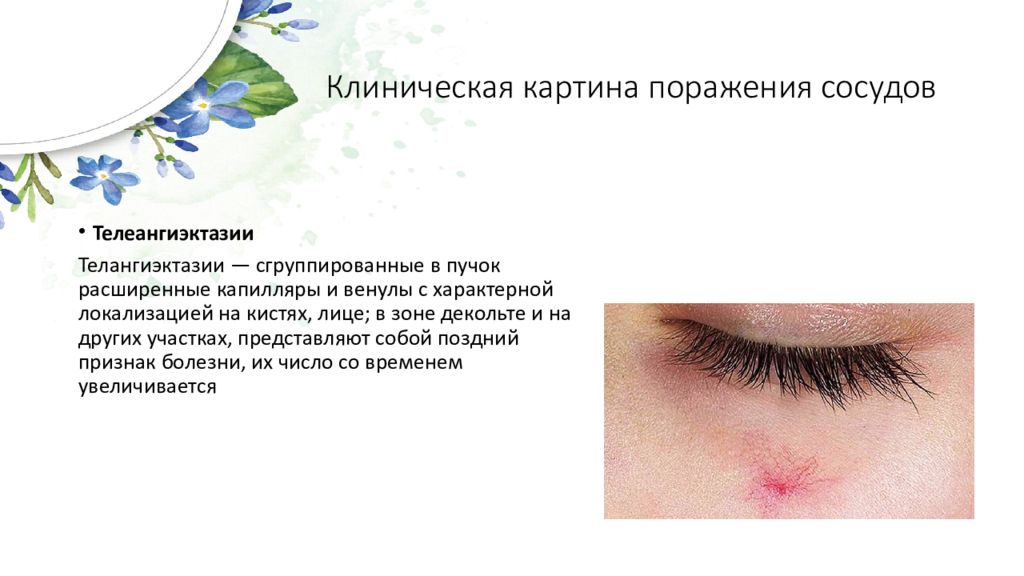 Активные очаги характеризуются сиреневым гало (лиловое кольцо), окружающим фиброзированный центр. По мере прогрессирования заболевания склеротические очаги вновь размягчаются, становятся атрофичными, гипо-/гиперпигментированными. В зависимости от локализации фиброза, заболевание также может ассоциироваться с потерей придатков кожи в пораженной области.
Активные очаги характеризуются сиреневым гало (лиловое кольцо), окружающим фиброзированный центр. По мере прогрессирования заболевания склеротические очаги вновь размягчаются, становятся атрофичными, гипо-/гиперпигментированными. В зависимости от локализации фиброза, заболевание также может ассоциироваться с потерей придатков кожи в пораженной области.
Каплевидная форма локализованной склеродермии характеризуется образованием небольших желтовато-белых склеротических очагов с блестящей поверхностью, возникающих вначале на коже туловища (диаметром менее 1 см, окруженных лиловым кольцом в период клинической активности). На ранних стадиях заболевания очаги могут иметь вид эритематозных пятен. Атрофодермия Пазини-Пьерини, возможно, является абортивной формой каплевидной склеродермии. Часто возникая в детском возрасте, данный вариант характеризуется появлением эритематозных очагов, диаметром менее 1 см, расположенных на туловище симметрично. Наблюдается некоторая депрессия очагов, вследствие потери соединительной ткани. В таких случаях гистологические изменения соответствуют поздней атрофической стадии локализованной склеродермии.
В таких случаях гистологические изменения соответствуют поздней атрофической стадии локализованной склеродермии.
Табл.1 Классификация локализованной склеродермии
1. Ограниченная форма
1) Морфеа (бляшечная форма)
2) Каплевидная морфеа (особая форма бляшечной склеродермии)
3) Атрофодермия Пазини-Пьерини(особая форма склеродермии)
2. Генерализованная форма
1) Генерализованная ограниченная склеродермия (с поражением минимум 3х анатомических областей)
2) Пансклеротическая морфеа (особая тяжелая форма)
3) Эозинофильный фасциит (особая форма с преимущественным поражением фасции)
3. Линейная форма
1) Линейная локализованная склеродермия (с преимущественным поражением конечностей)
2) Линейная локализованная склеродермия en coup de sabre
3) Прогрессируюащя гемиатрофия лица Парри-Ромберга
4. Глубокая форма
5. Смешанная форма
Комментарии: Ограниченные подтипы, поражающие исключительно кожу, включают бляшечную форму (классическая морфеа), каплевидную морфеа, атрофодерми Пазини-Пьерини. Подтипы с внекожными проявлениями включают линейную форму локализованной склеродермии (подтипы en coup de sabre, прогрессирующая гемиатрофия лица ), генерализованную форму (пансклеротическая склеродермия, эозинофильный фасциит), а также глубокую форму.
Подтипы с внекожными проявлениями включают линейную форму локализованной склеродермии (подтипы en coup de sabre, прогрессирующая гемиатрофия лица ), генерализованную форму (пансклеротическая склеродермия, эозинофильный фасциит), а также глубокую форму.
По мнению авторов, эозинофильный фасциит явялется особой формой локализованной склеродермии, которую можно отнести как к генерализованной, так и линейной форме.
Синонимом глубокой склеродермии является глубокая морфеа.
Смешанные варианты наиболее часто наблюдаются у детей. Часто сочетается линейная и бляшечная форма склеродермии либо линейная и генерализованная форма.
Генерализованная форма локализованной склеродермии
Этот вариант диагностируется при поражении трех и более анатомических зон. Типичная локализация — кожа туловища, бедра, люмбосакральная область. Представленные на разных стадиях развития заболевания очаги часто располагаются симметрично, могут сливаться в более крупные бляшки.
Крайне редкий вариант генерализованной склеродермии – пансклеротическая склеродермия представляет собой комбинацию линейной и диссеминированной формы локализованной склеродермии, характеризуется вовлечением обширных участков кожи с незначительной тенденцией к регрессу фиброза. Заболевание часто приводит к образованию выраженных контрактур, нарушению заживления ран, в некоторых случаях образуются обширные язвы.
Заболевание часто приводит к образованию выраженных контрактур, нарушению заживления ран, в некоторых случаях образуются обширные язвы.
Признаваемый многими экспертами особой формой локализованной склеродермии, эозинофильный фасциит (синдром Шульмана), по нашему мнению, является вариантом генерализованной склеродермии. Клинически данная форма представляет собой прогрессирующий фиброз проксимальных участков конечностей с различной степенью зпадения кожи (признак желоба, апельсиновой корки), обусловленный глубоко расположенным фиброзом в области фасции и подкожных перегородок. Заболевание часто провоцируется травматизацией, характеризуется эозинофилией крови и тканей.
Линейная форма локализованной склеродермии
Линейная форма локализованной склеродермии характеризуется появлением линейных полосовидных очагов, в некоторых случаях очаги генерализованные. При мягких формах очаги разрешаются с образованием гиперпигментации, при более тяжелых формах в случае расположения очагов в проекции суставов, образуются жесткие полосовидные очаги склероза, приводящие к значительному нарушению подвижности. В патологический процесс также могут вовлекаться мышцы, костная ткань в проекции очага на коже. Наиболее известный вариант линейной склеродермии — en coup de sabre, чаще всего очаг распространяяется от линии бровей до кожи скальпа (более характерна парамедианная локализация), сопровождается развитием рубцовой алопеции. В ряде случаев наблюдается вовлечение в процесс нервных волокон.
В патологический процесс также могут вовлекаться мышцы, костная ткань в проекции очага на коже. Наиболее известный вариант линейной склеродермии — en coup de sabre, чаще всего очаг распространяяется от линии бровей до кожи скальпа (более характерна парамедианная локализация), сопровождается развитием рубцовой алопеции. В ряде случаев наблюдается вовлечение в процесс нервных волокон.
Прогрессирующая гемиатрофия лица (синдром Парри-Ромберга) – состояние, близкое к линейной локализованной склеродермии. Крайне редкое заболевание, характеризующееся первичной атрофией подкожной ткани, мышц, костей, в некоторых случаях сопровождается гиперпигментацией кожи в очаге. Фиброз развивается редко. Часто начинаясь с небольшго очага в области головы у детей и подростков (см. «Характерные черты в детском возрасте»), заболевание прогрессирует, вовлекая мышцы щек, костей, язык, приводит к выраженной ассиметрии лица. Одновременное возникновение линейной склеродермии по типу en coup de sabre и прогрессирующей гемиатрофии лица описывается до 40% случаев. По этой причине в предложенной авторами классификации прогрессирующая гемиатрофия лица расположена в группе линейной локализованной склеродермии, однако, справедливо также будет отнести ее к глубокой локализованной склеродермии, так как наблюдается вовлечение в патологический процесс более глубоких структур. Вовлечение ЦНС не является редкостью. Антинуклеарные антитела могут обнаруживаться в 50% случаев.
По этой причине в предложенной авторами классификации прогрессирующая гемиатрофия лица расположена в группе линейной локализованной склеродермии, однако, справедливо также будет отнести ее к глубокой локализованной склеродермии, так как наблюдается вовлечение в патологический процесс более глубоких структур. Вовлечение ЦНС не является редкостью. Антинуклеарные антитела могут обнаруживаться в 50% случаев.
Глубокая форма локализованной склеродермии
Глубокая форма локализованной склеродермии – наиболее редкий вариант заболевания (менее 5 % случаев). В данном случае фибротический процесс первично поражает глубокие компоненты соединительной ткани – жировую ткань, фасции, подлежащие мышечные структуры. Преобладающая локализация – конечности, отдельные очаги часто расположены симметрично. Глубокая морфеа может возникать в детском возрасте, часто без предшествующего воспаления.
Смешанная форма
В ряде случаев поражение кожи нельзя отнести лишь к одному клиническому варианту. Локализованная склеродермия у детей часто проявляется сочетанием линейной и бляшечной склеродермии либо линейной и генерализованной склеродермии.
Локализованная склеродермия у детей часто проявляется сочетанием линейной и бляшечной склеродермии либо линейной и генерализованной склеродермии.
Классификация локализованной склеродермии включает следующие пять основных вариантов: ограниченная, генерализованная, линейная, глубокая и смешанная. Помимо основных, существует еще несколько подтипов(табл. 1).
Ассоциация с другими аутоиммунными заболеваниями
Взаимосвязь локализованной склеродермии с иными аутоиммунными заболеваниями известна на протяжении многих лет. В исследовании 245 пациентов с локализованной склеродермией , опубликованном в 2009г, 17,6% пациентов имели сопутствующие ревматические или аутоиммунные заболевания(в четыре раза чаще, чем в общей популяции), частота встречаемости у взрослыхпациентов была выше. Пациенты с генерализованной склеродермией намного чаще имеют сопутствующие аутоиммунные заболевания (45,9%, в 12 раз чаще, чем в общей популяции) по сравнению с другими формами локализованной склеродермии (9,6%). Наиболее часто локализованная склеродермия ассоциируется с бляшечным псориазом, системной красной волчанкой, рассеянным склерозом, витилиго. Около 16,3% пациентов имеют отягощенный семейный анамнез (по аутоиммунным заболеваниям), причем чаще ассоциация наблюдается у детей(23,8%),реже — у взрослых (10,6%). В ретроспективном исследовании 472 пациентов с локализованной склеродермией у 8,1% были обнаружены ассоциированные аутоиммунные заболевания – тиреоидит Хашимото, ревматоидный артрит, очаговая алопеция, СД I типа.
Наиболее часто локализованная склеродермия ассоциируется с бляшечным псориазом, системной красной волчанкой, рассеянным склерозом, витилиго. Около 16,3% пациентов имеют отягощенный семейный анамнез (по аутоиммунным заболеваниям), причем чаще ассоциация наблюдается у детей(23,8%),реже — у взрослых (10,6%). В ретроспективном исследовании 472 пациентов с локализованной склеродермией у 8,1% были обнаружены ассоциированные аутоиммунные заболевания – тиреоидит Хашимото, ревматоидный артрит, очаговая алопеция, СД I типа.
Ассоциация локализованной склеродермии и склероатрофического лихена, как правило, описывается в клинических наблюдениях и небольших сериях клинических случаев, наиболее часто наблюдается ассоциация с экстрагенитальным склероатрофическим лихеном. В проспективном исследовании, проведенном во Франции, из 76 пациентов, описанных в 2012г, 38% также имели генитальный склероатрофический лихен. Наиболее часто ассоциация наблюдалась у пациентов с бляшечной и генерализованной склеродермией. Впоследствии, ретроспективное исследование, выполненное в Германии, также подтвердило преобладание генитального склероатрофичского лихена у пациентов с лкоализованной склеродермией.
Впоследствии, ретроспективное исследование, выполненное в Германии, также подтвердило преобладание генитального склероатрофичского лихена у пациентов с лкоализованной склеродермией.
При наличии у пациента локализованной склеродермии необходимо исключить другие аутоиммунные и ревматические заболевания путем тщательного сбора анамнеза и осмотра пациента. При наличии подозрений необходимо дальнейшее дообследование.
- ! Также, необходимо исключить генитальный склероатрофический лихен путем осмотра аногенитальной области, особенно у пациентов с бляшечной и генерализованной локализованной склеродермией.
Патогенез
Патогенез заболевания детально описывается в приложении.
Лабораторные данные
Частые серологические изменения
В отличие от системной склеродермии, при которой наблюдаются высокоспецифичные антитела — anti-Scl-70 при диффузной форме (антитела к топоизомеразе 1) и антицентромерные антитела при ограниченной форме, при локализованной склеродермии характерных серологических изменений не наблюдается. По мнению авторов, помимо базовых лабораторных тестов (клинический анализ крови, биохимический анализ крови, антинуклеарные антитела), необходимо выполнение специальных лабораторных исследований для исключения системной склеродермии (антитела к топоизомеразе-1, антицентромерные антитела). (табл 2). В случае эозинофильного фасциита необходимо выполнение электрофореза и качественного определения иммуноглобулинов крови.
По мнению авторов, помимо базовых лабораторных тестов (клинический анализ крови, биохимический анализ крови, антинуклеарные антитела), необходимо выполнение специальных лабораторных исследований для исключения системной склеродермии (антитела к топоизомеразе-1, антицентромерные антитела). (табл 2). В случае эозинофильного фасциита необходимо выполнение электрофореза и качественного определения иммуноглобулинов крови.
Необходимо мониторирование активности заболевания. В случае эозинофильного фасциита оценивается уровень эозинофилов крови. Специфических маркеров активности локализованной склеродермии на сегодняшний день не установлено, оценка активности заболевания производится на основе клинической картины. Недавнее исследование показало, что повышение уровня креатинкиназы и альдолазы может указывать на активность процесса в случае ювенильной локализованной склеродермии,таким образом, в ряде случаев эти параметры могут быть полезны.
При ограниченной и глубокой локализованной склеродермии часто не обнаруживается серологических изменений. Напротив, линейная локализованная склеродермия, особенно у детей, в активную фазу часто сопровождается наличием антинуклеарных антител, антител к гистонам, гипергаммаглобулинемией, эозинофилией. По мнению авторов, обнаружение таких антител при локализованой склеродермии является сопутствующим симптомом неуточненной патогенетической значимости. В случае вовлечения суставов при линейной локализованной склеродермии на конечностях, может быть повышен ревматоидный фактор, что требует дальнейшего обследования для выявления пораженных суставов. Активная ранняя фаза эозинофильного фасциита характеризуется эозинофилией периферической крови, в некоторых случаях – значительно увеличенным СОЭ.
Напротив, линейная локализованная склеродермия, особенно у детей, в активную фазу часто сопровождается наличием антинуклеарных антител, антител к гистонам, гипергаммаглобулинемией, эозинофилией. По мнению авторов, обнаружение таких антител при локализованой склеродермии является сопутствующим симптомом неуточненной патогенетической значимости. В случае вовлечения суставов при линейной локализованной склеродермии на конечностях, может быть повышен ревматоидный фактор, что требует дальнейшего обследования для выявления пораженных суставов. Активная ранняя фаза эозинофильного фасциита характеризуется эозинофилией периферической крови, в некоторых случаях – значительно увеличенным СОЭ.
Значение Borrelia burgdorferi в патогенезе локализованной склеродермии остается противоречивым. По мнению авторов, на сегодняшний день нет убедительных данных в пользу наличия прямых патогенетических связей между Borrelia burgdorferi и локализованной склеродермией. Необходимость рутинной серологической диагностики Borrelia burgdorferi остается противоречивой.
Необходимость рутинной серологической диагностики Borrelia burgdorferi остается противоречивой.
Серологические параметры дальнейшего наблюдения за пациентом
На сегодняшний день параметры для оценки активности заболевания и мониторирования течения заболевания не разработаны для рутинной клинической практики.
- ! При всех формах локализованной склеродермии необходимо выполнять базовые лабораторные исследования (клинический анализ крови, биохимический анализ крови) и антинуклеарные антитела.
- ! Скрининг на наличие антител к экстрагируемому ядерному антигену должен проводиться только при наличии подозрения на иные аутоиммунные заболевания.
- ! Необходима разработка показателей активности заболевания
- ! Серологическое исследование на Borrelia burgdorferi должно производиться в отдельных обдуманных случаях.
Базовые и специальные лабораторные исследования при локализованной склеродермии
1. Клинический анализ крови с подсчетом клеток (особенно важно при эозинофильном фасциите и линейной склеродермии)
Клинический анализ крови с подсчетом клеток (особенно важно при эозинофильном фасциите и линейной склеродермии)
2. Биохимический анализ крови
1) Трансаминазы (АЛТ, АСТ)
2) Параметры холестаза (ГГТП)
3) ЛДГ
4) Креатинин
5) Креатинкиназа(особенно при подозрении на миозит)
6) СОЭ, СРБ
7) Антинуклеарные антитела (на колонии HEp-2 клеток )
Последующие исследования
1. Скрининг на антитела к ядерному экстрагируемому антигену (только при необходимости исключить системную склеродермию), также анализ крови на anti-Scl-70, антицентромерные антитела, антитела к гистонам часто обнаруживаются при линейной склеродермии с поражением конечностей у детей.
2. Ревматоидный фактор.
Гистологическое исследование
Биописия и фиксация материала
С учетом того, что локализованная склеродермия( в зависимости от клинического варианта) может распространяться вглубь подлежащих тканей, вплоть до фасции и мышц, необходимо выполнение достаточно глубокой биопсии. Как правило, достаточно стандартной фиксации в формалине.
Как правило, достаточно стандартной фиксации в формалине.
Гистология локализованной склеродермии
Локализованную склеродермию можно подразделить на воспалительную (ранняя фаза) и склеротическую (поздняя фаза). К примеру, типичный очаг в виде бляшки представляет собой центральный склероз с воспалительным венчиком по периферии в виде лилового кольца в ранную фазу. Гистологические изменения при локализованной склеродермии могут быть весьма схожи с системной склеродермией, таким образом, микроскопия гистологического препарата не может служить достоверным методом для дифференциальной диагностики локализованной и системной склеродермии. Гистологическая картина должна оцениваться в соответствии с клинической картиной.
В раннюю фазу в сетчатом слое дермы обнаруживается плотный периваскулярный и периаднексальный инфильтрат, в некоторых случаях, в зависимости от конкретного клинического случая – инфильтрат распространяется в подкожные ткани. Инфильтрат представлен преимущественно лимфоцитами, часто присутствуют плазматические клетки и гистиоциты, также, могут обнаруживаться эозинофилы. Было показано, что при ювенильных формах отсутствуют дермальные CD 34+ дендритные клетки, в то время как уровень фактор XIIIa1-дермальных дендроцитов в очагах фиброза повышен. Соединительная ткань дермы часто содержит местами утолщенные пучки коллагеновых волокон (расположенные параллельно), в верхней части дермы обнаруживается отек. Поздняя фаза характеризуется склерозом дермы с уменьшением числа придаточных структур. Эккринные потовые железы атрофичны и «замурованы» вновь образованными волокнами коллагена. Вследствие утолщения кожи и вовлечения в процесс подкожной жировой клетчатки они расположены выше в дерме. Стенки мелких кровеносных сосудов также утолщены. Воспалительный инфильтрат, как правило, не выражен, коллагеновые волокна с выраженной эозинофилией плотно упакованы.
Было показано, что при ювенильных формах отсутствуют дермальные CD 34+ дендритные клетки, в то время как уровень фактор XIIIa1-дермальных дендроцитов в очагах фиброза повышен. Соединительная ткань дермы часто содержит местами утолщенные пучки коллагеновых волокон (расположенные параллельно), в верхней части дермы обнаруживается отек. Поздняя фаза характеризуется склерозом дермы с уменьшением числа придаточных структур. Эккринные потовые железы атрофичны и «замурованы» вновь образованными волокнами коллагена. Вследствие утолщения кожи и вовлечения в процесс подкожной жировой клетчатки они расположены выше в дерме. Стенки мелких кровеносных сосудов также утолщены. Воспалительный инфильтрат, как правило, не выражен, коллагеновые волокна с выраженной эозинофилией плотно упакованы.
Гистологическая картина эозинофильного фасциита
Ранняя фаза заболевания характеризуется плотным интерстициальным воспалительными инфильтратом, состоящим из моноцитов, плазматических клеток, эозинофилов. Эти инфильтраты распространяются из глубоких слоев дермы через подкожную жировую клетчатку вплоть до фасции и мышечной ткани. Воспалительная реакция приводит к выраженному утолщению фасции. Подобно приведенному ранее описанию, поздняя фаза в типичных случаях характеризуется наличием скудного воспалительного инфильтрата с выраженным безклеточным фиброзом, вовлекающим преимущественно фасцию. На данном этапе эозинофилы практически не обнаруживаются.
Эти инфильтраты распространяются из глубоких слоев дермы через подкожную жировую клетчатку вплоть до фасции и мышечной ткани. Воспалительная реакция приводит к выраженному утолщению фасции. Подобно приведенному ранее описанию, поздняя фаза в типичных случаях характеризуется наличием скудного воспалительного инфильтрата с выраженным безклеточным фиброзом, вовлекающим преимущественно фасцию. На данном этапе эозинофилы практически не обнаруживаются.
! В случае неясной клинической картины при локализованной склеродермии для подтверждения диагноза необходимо выполнение биопсии.(стандартная фиксация в формалине достаточна)
! При подозрении на глубокую, генерализованную и/или линейную форму, с учетом частого вовлечения глубоких структур, необходимо выполнять глубокую биопсию с захватом подкожной жировой клетчатки.
! При подозрении на эозинофильный фасциит необходимо выполнение глубокой биопсии с захватом фасции
Клиническая оценка (шкалы) и тактика диагностики
Радиологические процедуры
С учетом того, что неврологические симптомы, такие как мигрень и эпилепсия, чаще наблюдаются при линейной локализованной склеродермии, например, подтипе en coup de sabre и синдроме Парри-Ромберга, пациентам требуется проведение тщательного неврологического обследования, МРТ черепа для исключения вовлечения ЦНС.
Часто описывается субкортикальная кальцификация и атрофия головного мозга. Несмотря на то, что патологический процесс часто бывает асимптомным, поражение ЦНС может характеризоваться манифестацией различных неврологических симптомов. Так как дети часто не способны дать информации о возможных неврологических, суставных и/или глазных симптомах, необходимо совместное решение врача и пациента о необходимости выполнения радиологического исследования.
Более того, выполнение МРТ необходимо для планирования хирургического лечения (например, при подтипе en coup de sabre), а также как часть плана обследования при мышечных и костных симптомах, например, при линейной локализованной склеродермии.
К тому же, пациенты с линейной локализованной склеродермией (подтип en coup de sabre и синдром Парри-Ромберга) должны обследоваться офтальмологом, а также ортодонтом/ревматологом для исключения вовлечения в процесс височно-нижнечелюстного сустава.
Шкалы клинической оценки
Modified Localized Scleroderma Skin Severity Index (mLoSSI) явялется эквивалентом modified Rodnan Skin Score (mRSS) , используемого при системной склеродермии. По шкале от 0 до 3 производится оценка эритемы, утолщения кожи, а также появления новых очагов в 18 различных анатомических областях. Индекс может применяться как у взрослых, так и у детей. Дополнительный индекс, Localized Scleroderma Skin Damage Index (LoSDA) , был создан не только для оценки активных воспалительных очагов, но и эффекта от терапии. Оценивается дермальная и подкожная атрофия, депигментация, существующее повреждение кожи в 18 различных анатомических зонах по шкале от 0 до 3. Комбинация индексов LoSDA и Physician’s Global Assessment (PGA) обозначается как Localized Scleroderma Cutaneous Assessment Tool (LoSCAT). Этот валидизированный индекс позволяет иследователю раздельно оценивать активные и неактивные очаги, схожие индексы уже успешно используются при других ревматологических заболеваниях, например, CLASI или RCLASI при кожной красной волчанке. Использование LoSCAT, особенно в сочетании с mLoSSI, было положительно оценено при ювенильной локализованной склеродермии, таким образом, валидизированный индекс LoSCAT может успешно применяться для оценки течения заболевания как у взрослых, так и у детей. Использование индекса mLoSSI различными исследователями показало отличные результаты по отношению к эритеме и утолщению кожи. Несмотря на то, что наблюдается лишь умеренная корреляция между mLoSSI и PGA, оба варианта хорошо отражают ответ на терапию.
Этот валидизированный индекс позволяет иследователю раздельно оценивать активные и неактивные очаги, схожие индексы уже успешно используются при других ревматологических заболеваниях, например, CLASI или RCLASI при кожной красной волчанке. Использование LoSCAT, особенно в сочетании с mLoSSI, было положительно оценено при ювенильной локализованной склеродермии, таким образом, валидизированный индекс LoSCAT может успешно применяться для оценки течения заболевания как у взрослых, так и у детей. Использование индекса mLoSSI различными исследователями показало отличные результаты по отношению к эритеме и утолщению кожи. Несмотря на то, что наблюдается лишь умеренная корреляция между mLoSSI и PGA, оба варианта хорошо отражают ответ на терапию.
Диагностические методы для определения степени вовлечения кожи
Возможными методами, способными оценить степень вовлечения кожи являются 20-MHz ультразвук, Computerized Skin Score, кутометр, дурометр, термография, лазерное допплеровское исследование.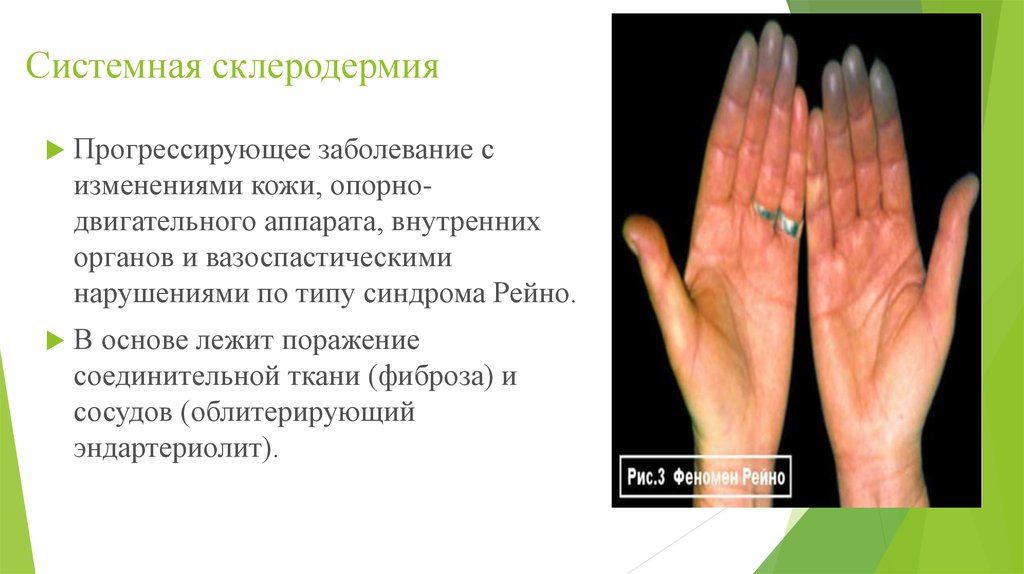 Эти методы достаточно длительно испоьзуются для оценки эффективности терапии в клинических исследованиях. Более детальная информация приведена в приложении.
Эти методы достаточно длительно испоьзуются для оценки эффективности терапии в клинических исследованиях. Более детальная информация приведена в приложении.
- ! При линейной локализованной склеродермии en coup de sabre и синдроме Парри-Ромберга требуется выполнение неврологического обследования и МРТ черепа для исключения вовлечения в процесс ЦНС.
- ! Возможными методами оценки глубины вовлечения кожи являются являются 20-MHz ультразвук, Computerized Skin Score, кутометр, дурометр, термография, лазерное допплеровское исследование.
- ! Предпочтительной шкалой для оценки локализованной склеродермии является валидизированная шкала LoSCAT.
Дифференциальная диагностика
С учетом различных фаз и клинического проявления заболевания существует широкий круг возможных диагнозов, которые необходимо принимать во внимание. В раннюю воспалительную фазу ограниченной склеродермии (морфеа), которая начинается с пятнистой, умеренно воспалительной, медленной увеличивающейся в размерах эритемы, дифференциальная диагностика проводится с ранней стадией экстрагенитального склероатрофического лихена, мигрирующей жритемой, кожным мастоцитозом, кольцевидной гранулемой, радиационным дерматитом, грибовидным микозом, а также реакцией на лекарственные препараты. В случае пигметной морфеа дифференциальный диагноз проводится с поствоспалительной гиперпигментацией, актиническим КПЛ, пятнами café-au-lait.
В случае пигметной морфеа дифференциальный диагноз проводится с поствоспалительной гиперпигментацией, актиническим КПЛ, пятнами café-au-lait.
Длительно протекающая морфеа может приводить к атрофии с потерей волос и сальных желез, что требует исключения хронического атрофического акродерматита(acrodermatitis chronica atrophicans), а также липодистрофии, склероатрофического лихена, рубцевания. В позднюю склеротическую фазу морфеа при локализации на нижних конечностях необходимо исключать липоидный некробиоз и претибиальную микседему.
Дифференциальная диагностика генерализованной склеродермии проводится со следующими заболеваниями: системная склеродермия, псевдосклеродермия? склередема взрослых, склеромикседема (papular mucinosis), склеродермоподобная форма РТПХ, смешанное заболевание соединительной ткани, нефрогенный системный фиброз. Дифференциальная диагностика линейной локализованной склеродермии en coup de sabre или прогрессирующей гемиатрофии лица Парри-Ромберга проводится с панникулитом, прогрессивной парциальной липодистрофией, фокальной дермальной гипоплазией, кортикостероид-индуцированной атрофией, глубокой красной волчанкой (люпус-панникулит). В таблице 3 приводится обзор заболеваний, с которыми требуется проводить дифференциальную диагностику.
В таблице 3 приводится обзор заболеваний, с которыми требуется проводить дифференциальную диагностику.
Таблица 3. дифференциальная диагностика локализованной склеродермии.
1. Ранняя воспалительная фаза локализованной склеродермии (морфеа)
1) Склероатрофический лишай
2) Мигрирующая эритема
3) Кожный мастоцитоз
4) Кольцевидная гранулема
5) Радиационный дерматит
6) Грибовидный микоз
7) Лекарственная реакция
2. Поздняя стадия ограниченной склеродермии (морфеа) с гиперпигментацией
1) Поствоспалительная гиперпигментация
2) Актинический КПЛ
3) Пятна цвета «кофе с молоком»
4) Erythema dyschromicum perstans
3. Поздняя стадия ограниченной склеродермии (морфеа) с атрофией
1) Acrodermatitis chronica atrophicans
2) Липодистрофия
3) Склероатрофический лишай
4) Рубец
4. Генерализованная склеродермия
1) Системная склеродермия
2) Псевдосклеродермия
3) Склередема взрослых Бушке
4) Склеромикседема (папулярный муциноз)
5) Склеродермоподобная форма РТПХ
6) Смешанное заболевание соединительной ткани
7) Нефрогенный системный фиброз
5. Линейная локализованная склеродермия
Линейная локализованная склеродермия
1) Панникулит
2) Прогрессивная парциальная липодистрофия
3) Фокальная дермальная гипоплазия
4) Кортикостероид-индуцированная атрофия
5) Глубокая красная волчанка (волчаночный панникулит)
Лечение локализованной склеродермии
Несмотря на то, что на сегодняшний день не существует метода лечения, направленного на устранение причины заболевания, разработаны достаточно эффективные терапевтические подходы, особенно в активную фазу заболевания. Так как различные подтипы локализованной склеродермии поражают различные ткани, авторы разработали алгоритм лечения, основанный на распространенности, тяжести процесса и подтипе заболевания. (Табл. 4) Важность такого подхода заключается в том, что некоторые формы бляшечной локализованной склеродермии часто не вызывают у пациента субъективных симптомов и беспокоят исключительно как косметический дефект. С другой стороны, выраженные формы линейной локализованной склеродермии в детском возрасте могут приводить к тяжелым стойким физическим и психологическим нарушениям, поэтому требуют раннего назначения системной терапии. В последующих параграфах будут описаны и оценены различные терапевтические подходы к лечению локализованной склеродермии. Эти рассуждения составляют основу упомянутой ранее концепции лечения. При оценке эффективности различных методов лечения необходимо помнить, что в большинстве случаев клиническое улучшение начинается не ранее 8 -12 недели терапии.
В последующих параграфах будут описаны и оценены различные терапевтические подходы к лечению локализованной склеродермии. Эти рассуждения составляют основу упомянутой ранее концепции лечения. При оценке эффективности различных методов лечения необходимо помнить, что в большинстве случаев клиническое улучшение начинается не ранее 8 -12 недели терапии.
Топическая терапия
Топические/внутриочаговые ГКС
Исследований по изучению эффективности тГКС при лечении локализованной склеродермии не проводилось. Однако, по мнению авторов, тГКС особенно эффективны в активную фазу поверхностных форм локализованной склеродермии, например морфеа. Для лечения должны использоваться очень сильные тГКС в течение 1 месяца, либо умеренные в течение 3 месяцев, аппликации производятся 1 раз в день. Нанесение под окклюзию может повысить эффективность препарата. Длительное применение тГКС должно проводиться курсами, в виде интервальной терапии. Внутриочаговое введение тГКС производится только в активную границу редкого подтипа en coup de sabre . Наиболее часто используется триамцинолона ацетонид в дозе 10-40 мг, в чистом виде, либо разведенный с лидокаином (от 1:2 до 1:4). Исследований по данному методу терапии не проводилось.
Наиболее часто используется триамцинолона ацетонид в дозе 10-40 мг, в чистом виде, либо разведенный с лидокаином (от 1:2 до 1:4). Исследований по данному методу терапии не проводилось.
Топический кальципотриол
Помимо клинических наблюдений было проведено два исследования по эффективности использования топического кальципотриола в дозе 0,005% при локализованной склеродермии. В одном из этих исследований кальципотриол в дозе 0,005% сочетался с проведением низкодозной UVA1-фототерапии. Аппликации кальципотриола проводились два раза в день, в исследовании с применением препарата в качестве монотерапии кальципотриол наносился под окклюзию. По мнению авторов, кальципотриол является достаточно эффективным препаратом, особенно в случае бляшечной склеродермии (морфеа). Препарат необходимо наносить дважды в день, минимум 3 месяца, по возможности под окклюзию.
Топические ингибиторы кальциневрина
Два пилотных исследования и несколько клинических наблюдений описывают успешное применение такролимуса в дозе 0,1% под окклюзию при морфеа. После 3 месяцев применения все пациенты отметили регресс эритемы и уменьшение склероза. В 2009г эти данные были подтверждены рандомизированным двойным слепым исследованием у 10 пациентов. По мнению авторов, с учетом полученных данных, мазь такролимус 0,1% может использоваться в качестве альтернативы топическим глюкокортикоидам в активную фазу заболевания. По данным авторов, исследований по поводу эффективности топического пимекролимуса не было опубликовано.
После 3 месяцев применения все пациенты отметили регресс эритемы и уменьшение склероза. В 2009г эти данные были подтверждены рандомизированным двойным слепым исследованием у 10 пациентов. По мнению авторов, с учетом полученных данных, мазь такролимус 0,1% может использоваться в качестве альтернативы топическим глюкокортикоидам в активную фазу заболевания. По данным авторов, исследований по поводу эффективности топического пимекролимуса не было опубликовано.
Имиквимод
Имиквимод – топический модификатор иммунного ответа — ингибирует трансформирующий фактор роста-бета (TGF-β) путем индукции интерферона гамма (IFN-γ), таким образом проявляя антифибротический эффект. Несмотря на то, что использование имиквимода при локализованной склеродермии представляется рациональным, на сегодняшний день существует весьма ограниченное количество клинических наблюдений, небольшой обзор серии клинических случаев и проспективное исследование опубликованное в 2011г. В последнее были включены 9 детей с морфеа, которые получали терапию имиквимодом 5% в течение 9 месяцев, что привело к значительному уменьшению толщины дермы после проведенного лечения. Собственный опыт авторов не является настолько позитивным. Основываясь на полученных данных, имиквимод не может быть рекомендован в качестве терапии локализованной склеродермии.
Собственный опыт авторов не является настолько позитивным. Основываясь на полученных данных, имиквимод не может быть рекомендован в качестве терапии локализованной склеродермии.
Внутриочаговое введение IFN-γ
Эффективность внутриочагового введения IFN-γ была исследована в двойном слепом плацебо контролируемом исследовании. По сравнению с плацебо выраженного терапевтического эффекта не наблюдалось. Таким образом, использование внутриочагового введения IFN-γ при лечении локализованной склеродермии не рекомендуется.
- ! В активную фазу заболевания при лечении локализованной склеродермии с ограниченным поражением кожи могут применяться однократные ежедневные аппликации топических глюкокортикоидов умеренной или высокой активности(сильные тГКС – 4 недели, умеренной силы – 12 недель). Для повышения эффективности могут проводиться аппликации препарата под окклюзию. Длительная терапия ГКС должна проводиться в виде пульс-терапии.
- ! В качестве альтернативы может использоваться топический кальципотриол или топические ингибиторы кальциневрина.
Фототерапия
Терапия с использованием УФ-излучения признана одной из наиболее эффективных методик при склерозирующих заболеваниях кожи. В основе действия УФ-излучения лежит способность индуцировать интерстициальную коллагеназу (матриксную металлопротеиназу-1) in vitro. Вдохновленные этими находками, последующие эксперименты показали, что облучение здоровой кожи при помощи длинноволнового УФ-А излучения также индуцирует интерстициальную коллагеназу. Лечение с использованием ультрафиолетового облучения обладает противовоспалительным и антифибротическим эффектом. Оно приводит к апоптозу дермальных Т-клеток, деплеции клеток Лангерганса, модификации ряда провоспалительных цитокинов. Как было описано выше, антифибротический эффект основывается на индукции различных матриксных металлопротеиназ и ингибировании продукции коллагена.
В течение последних 20 лет было опубликовано значительное количество ключевых статей по лечению склерозирующих заболеваний кожи при помощи фототерапии.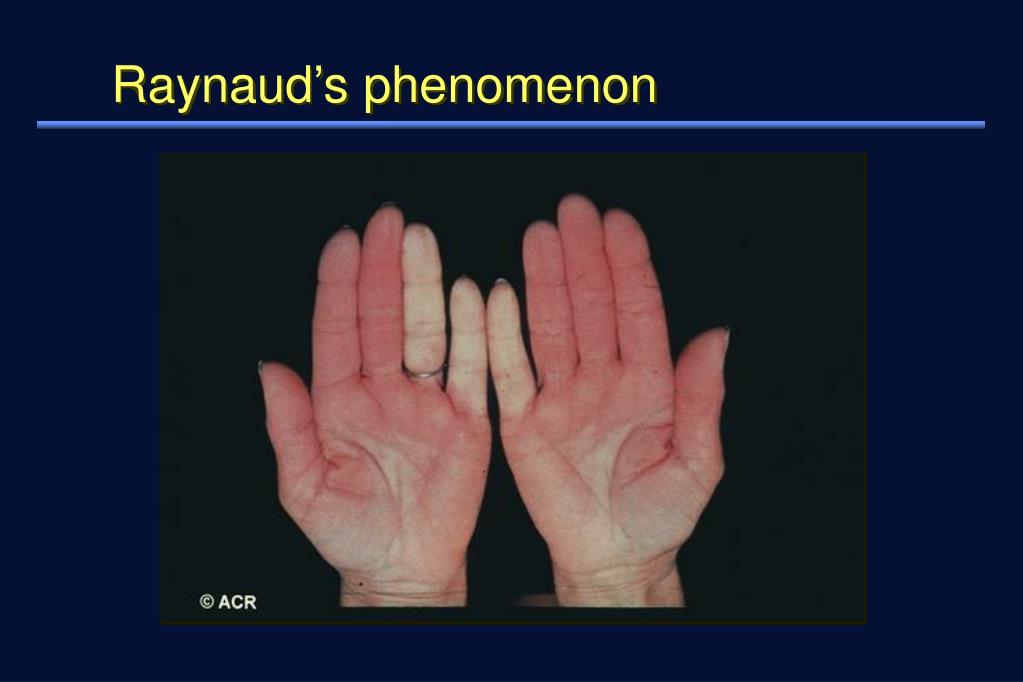 Длинноволновое УФ-излучение достигает глубоких слоев дермы. Таким образом, по мнению авторов, УФ-терапия является терапией первой линии при ограниченной локализованной склеродермии. Напротив, УФ-терапия не подходит для лечения глубоких форм (с вовлечением подкожной жировой клетчатки, фасции, мышечной ткани, костной ткани).
Длинноволновое УФ-излучение достигает глубоких слоев дермы. Таким образом, по мнению авторов, УФ-терапия является терапией первой линии при ограниченной локализованной склеродермии. Напротив, УФ-терапия не подходит для лечения глубоких форм (с вовлечением подкожной жировой клетчатки, фасции, мышечной ткани, костной ткани).
ПУВА-терапия
При проведении ПУВА-терапии используются фотосенсибилизаторы псоралены, которые могут приниматься пациентом перорально (системная ПУВА-терапия), либо наноситься на кожу в виде аппликаций (локальная ПУВА-терапия либо ПУВА-ванны). В Германии наиболее распространено лечение 8-метоксипсораленом. При проведении ПУВА-ванн пациент принимает 20-минутную ванну в воде, содержащей 8-метоксипсорален, при температуре 37 градусов Цельсия. Затем производится облучение широкополосным УФ-А излучением. Начальная доза УФ-А излучения применяется после определения индивидуальной минимальной фототоксической дозы. Так как максимум развития ПУВА-индуцированной эритемы приходится на 72 часа после облучения, минимальная фототоксическая доза оценивается не ранее этого срока. Системная ПУВА-терапия начинается с дозы, составляющей 50-70% от минимальной фототоксической дозы. При локальной ПУВА-терапии и ПУВА-ваннах начальная доза составляет 30% от минимальной фототоксической дозы. При локализованной склеродермии предпочтение отдается ПУВА-ваннам, так как этот вариант терапии не сопровождается побочными явлениями со стороны ЖКТ, характерными для перорального приема 8-метоксипсоралена. Помимо нескольких клинических наблюдений существует два ретроспективных исследования. Более крупное исследование было опубликовано в 2013г, включало 28 пациентов (23 женщины и 5 мужчин), получавших ПУВА-ванны 3 раза в неделю. Полное разрешение процесса наблюдалось у 39% пациентов, в 50% случаев отмечалась положительная динамика процесса, 10% — без ответа на терапию. Сравнительно неплохие результаты были получены при проведении локальной ПУВА-терапии у 4 пациентов с локализованной склеродермией. Требуется проведение дальнейших исследований. ПО мнению авторов, ПУВА-ванны могут быть особенно эффективны при использовании в раннюю воспалительную фазу ограниченной локализованной склеродермии.
Системная ПУВА-терапия начинается с дозы, составляющей 50-70% от минимальной фототоксической дозы. При локальной ПУВА-терапии и ПУВА-ваннах начальная доза составляет 30% от минимальной фототоксической дозы. При локализованной склеродермии предпочтение отдается ПУВА-ваннам, так как этот вариант терапии не сопровождается побочными явлениями со стороны ЖКТ, характерными для перорального приема 8-метоксипсоралена. Помимо нескольких клинических наблюдений существует два ретроспективных исследования. Более крупное исследование было опубликовано в 2013г, включало 28 пациентов (23 женщины и 5 мужчин), получавших ПУВА-ванны 3 раза в неделю. Полное разрешение процесса наблюдалось у 39% пациентов, в 50% случаев отмечалась положительная динамика процесса, 10% — без ответа на терапию. Сравнительно неплохие результаты были получены при проведении локальной ПУВА-терапии у 4 пациентов с локализованной склеродермией. Требуется проведение дальнейших исследований. ПО мнению авторов, ПУВА-ванны могут быть особенно эффективны при использовании в раннюю воспалительную фазу ограниченной локализованной склеродермии.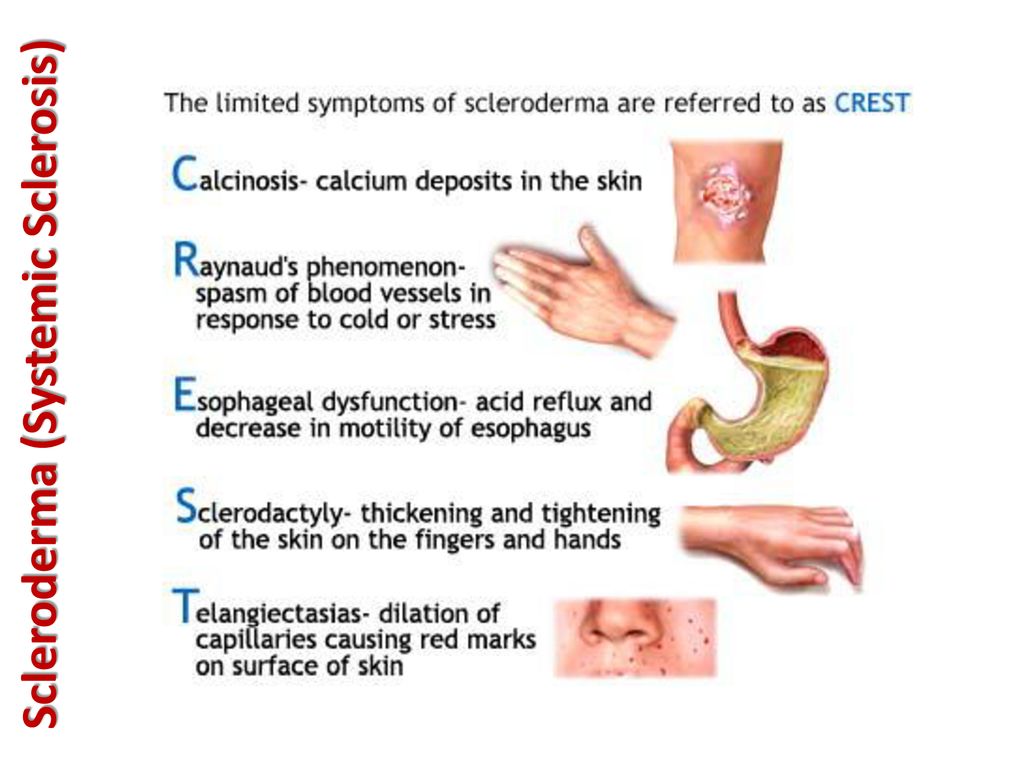 Цикл терапии должен включать около 30 сеансов, проводимых 2-3 раза в неделю.
Цикл терапии должен включать около 30 сеансов, проводимых 2-3 раза в неделю.
Авторы не рекомендуют проведение бальнеотерапии при локализованной склеродермии, так как отсутствуют данные об ее эффективности.
Широкополосная UVA-фототерапия
На сегодняшний день опубликовано 3 проспективных исследования, посвященных использованию широкополосной UVA-фототерапии (320-400 нм) при локализованной склеродермии, в самом крупном исследовании участвовало 63 пациента. Схожие результаты были получены при применении трех дозировок – 5, 10 и 20 Дж/ cм² курсом из 20 процедур (кумулятивная доза 100, 200 и 400 Дж/cм² ). Таким образом, низкодозная терапия представляется эквивалентной по эффективности более высоким дозам. На сегодняшний день, контролируемых исследований по изучению широкополосной UVA-фототерапию, не проводилось, ее эффективность не сравнивалась с иными вариантами фототерапии. По опыту авторов рекомендаций, широкополосная UVA-фототерапия представляется менее эффективной по сравнению с ПУВА-терапией и UVA1-фототерапией и должна применяться лишь при отсутствии доступа к ПУВА и UVA1.
UVA1-фототерапия
Ключевым моментом в развитии UVA1-фототерапии стало изобретение в 1981г радиационной лампы с шириной спектра 340-400 нм. шириной спектра 340-400 нм. Наиболее часто используются лампы с пиком эмиссии около 370 нм. Различают три режима дозирования: низкодозная UVA1-фототерапия (10-20 Дж/cм²), UVA1-фототерапия средней дозы (30-50 Дж/cм²) и высокодозная UVA1-фототерапия(60-130 Дж/cм²). В терапии локализованной склеродермии применяются все три режима. В первом проспективном исследовании UVA1-фототерапии высокоэффективной оказалась высокодозная UVA1-фототерапия, в то время как выраженного эффекта при использовании низких доз не наблюдалось. В нескольких проспективных исследованиях, проведенных позднее, использоование низких и средних доз UVA1-фототерапии оказалось эффективным, однако, в исследованиях применялись преимущественно средние дозы. Единственное рандомизированное контролируемое исследование, посвященное UVA1-фототерапии, показало более высокую эффективность средних доз UVA1-фототерапии по сравнению с низкими дозами. Остается невыясненным вопрос – наблюдается ли худший ответ на UVA1-фототерапию у пациентов с темным фототипом кожи. Возникновение рецидива заболевания в течение 3х лет возможно у 50% пациентов. В таких случаях проводится дополнительный курс фототерапии. По мнению авторов рекомендаций, предпочтительными являются средние дозы UVA1-фототерапии (3-5 раз в неделю, суммарно около 40 сеансов).
Остается невыясненным вопрос – наблюдается ли худший ответ на UVA1-фототерапию у пациентов с темным фототипом кожи. Возникновение рецидива заболевания в течение 3х лет возможно у 50% пациентов. В таких случаях проводится дополнительный курс фототерапии. По мнению авторов рекомендаций, предпочтительными являются средние дозы UVA1-фототерапии (3-5 раз в неделю, суммарно около 40 сеансов).
! Терапией первой линиии при ограниченных подтипах локализованной склеродермии являются средние дозы UVA1-фототерапии. В качестве альтернативы может использоваться ПУВА-ванны либо локальная ПУВА-терапия.
Системная терапия
Системные ГКС
Ряд исследований продемонстрировал эффективность системных ГКС в острую стадию заболевания как в виде монотерапии, так и в комбинации с другими препаратами. ГСК использовались системно в более тяжелых случаях, при прогрессирующей генерализованной склеродермии или линейной склеродермии, подтип en coup de sabre. Исследование, в котором применялась монотерапия пероральными ГКС, преимущественно у взрослых пациентов, показало явное улучшение кожной симптоматики практически у всех пациентов, средняя продолжительность лечения составила 18 месяцев (дозировака 0,5-1,0 мг/кг ежедневно). Рецидив произошел у 1/3 пациентов после прекращения терапии ГКС. Эозинофильный фасциит, как правило, очень хорошо отвечает на терапию ГКС. У большинства пациентов с этим подтипом монотерапии системными ГКС бывает достаточно. В других исследованиях для лечения локализованной склеродермии применялась комбинация системных глюкокортикоидов с метотрексатом. По мнению авторов, с учетом развития возможынх побочных явлений, моноторапия системными глюкокортикоидами может проводиться кратковременно в раннюю острую фазу тяжелых форм локализованной склеродермии.
Исследование, в котором применялась монотерапия пероральными ГКС, преимущественно у взрослых пациентов, показало явное улучшение кожной симптоматики практически у всех пациентов, средняя продолжительность лечения составила 18 месяцев (дозировака 0,5-1,0 мг/кг ежедневно). Рецидив произошел у 1/3 пациентов после прекращения терапии ГКС. Эозинофильный фасциит, как правило, очень хорошо отвечает на терапию ГКС. У большинства пациентов с этим подтипом монотерапии системными ГКС бывает достаточно. В других исследованиях для лечения локализованной склеродермии применялась комбинация системных глюкокортикоидов с метотрексатом. По мнению авторов, с учетом развития возможынх побочных явлений, моноторапия системными глюкокортикоидами может проводиться кратковременно в раннюю острую фазу тяжелых форм локализованной склеродермии.
Метотрексат
Среди всех вариантов системной терапии наибольшее количество доступной информации относится к применению метотрексата. На сегодняшний день, проведено одно плацебо-контролируемое мультицентровое исследование, три проспективных и четыре ретроспективных исследования. Рандомизированное, двойное слепое, плацебо-контролируемое исследование включало 70 детей с локализованной склеродермией (46 пациентов в группе метотрексата и 24 в группе плацебо). Все пациенты с активным заболеванием были рандомизированы с соотнощением 2:1 и получали либо метотрексат перорально (15 мг/ м² один раз в неделю, максимальная доза 20 мг) либо плацебо в течение 12 месяцев. К тому же, обе группы получали преднизолон (1 мг/кг ежедневно, максимальная дооза 50 мг) в течение первых 3 месяцев. Для оценки параметров использовались специальные клинические шкалы (компьютеризированная система оценки, SSR ) и инфракрасная термография. В отличие от группы плацебо, группа метотрексата показала значительное уменьшение значений SSR (с 1 до 0,79), также как и температуры кожи при использовании инфракрасной термографии (снижение на 44%) после 12 месяцев. Уровень рецидивов в группе метотрексата был значительно ниже (32,6%) по сравнению с плацебо (70,8%). В трех проспективных исследованиях 34 пациента (24 взрослых и 10 детей) получали комбинированную терапию высокими дозами метилпреднизолона и метотрексатом (взрослые 15 мг в неделю, дети 0,3 мг/кг в неделю).
Рандомизированное, двойное слепое, плацебо-контролируемое исследование включало 70 детей с локализованной склеродермией (46 пациентов в группе метотрексата и 24 в группе плацебо). Все пациенты с активным заболеванием были рандомизированы с соотнощением 2:1 и получали либо метотрексат перорально (15 мг/ м² один раз в неделю, максимальная доза 20 мг) либо плацебо в течение 12 месяцев. К тому же, обе группы получали преднизолон (1 мг/кг ежедневно, максимальная дооза 50 мг) в течение первых 3 месяцев. Для оценки параметров использовались специальные клинические шкалы (компьютеризированная система оценки, SSR ) и инфракрасная термография. В отличие от группы плацебо, группа метотрексата показала значительное уменьшение значений SSR (с 1 до 0,79), также как и температуры кожи при использовании инфракрасной термографии (снижение на 44%) после 12 месяцев. Уровень рецидивов в группе метотрексата был значительно ниже (32,6%) по сравнению с плацебо (70,8%). В трех проспективных исследованиях 34 пациента (24 взрослых и 10 детей) получали комбинированную терапию высокими дозами метилпреднизолона и метотрексатом (взрослые 15 мг в неделю, дети 0,3 мг/кг в неделю).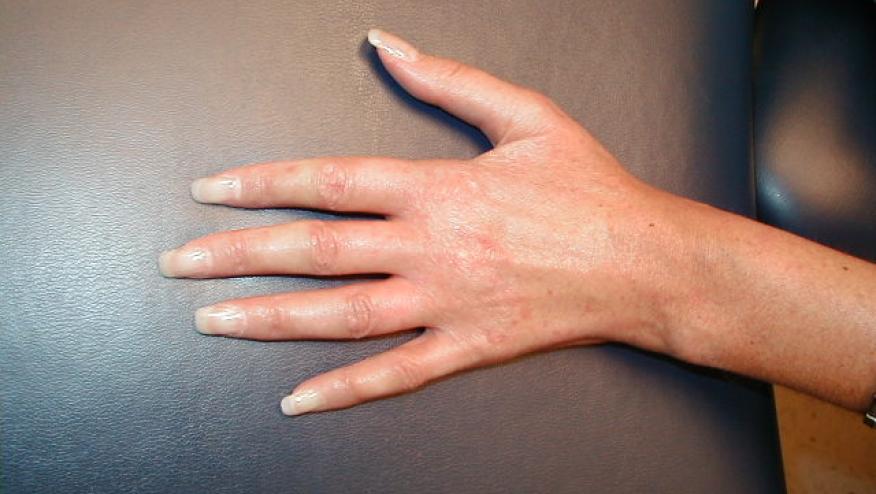 Для оценки течения заболевания использовалась невалидизированная клиническая шкала и ультразвуковое исследование кожи. Лечение привело к значительному улучшению у всех взрослых и у 9 из 10 детей. В четырех ретроспективных исследованиях терапию получали 119 пациентов (52 получали монотерапию метотрексатом, 67 пациентов – комбинированную терапию, метотрескат и системные ГКС, дозировка метотрексата: 15-20 мг в неделю у взрослых и 0,3 мг/кг в неделю у детей). Системные ГКС вначале вводились внутривенно, затем принимались перорально. Улучшение наблюдалось у 97% пациентов. Рецидив после завершения терапии в группе метотрексата составил 28%, в среднем через 1,7 года после завершения терапии. Режимы дозирования для комбинированной терапии метотрексат-системные ГКС: В приведенных выше исследованиях наблюдалась значительная вариация в размере использовавшихся доз. В 2012г CARRA (Childhood Arthritis and Rheumatology Research Alliance), впервые разработали три различных варианта терапии для лечения локализованной склеродермии у детей: 1.
Для оценки течения заболевания использовалась невалидизированная клиническая шкала и ультразвуковое исследование кожи. Лечение привело к значительному улучшению у всех взрослых и у 9 из 10 детей. В четырех ретроспективных исследованиях терапию получали 119 пациентов (52 получали монотерапию метотрексатом, 67 пациентов – комбинированную терапию, метотрескат и системные ГКС, дозировка метотрексата: 15-20 мг в неделю у взрослых и 0,3 мг/кг в неделю у детей). Системные ГКС вначале вводились внутривенно, затем принимались перорально. Улучшение наблюдалось у 97% пациентов. Рецидив после завершения терапии в группе метотрексата составил 28%, в среднем через 1,7 года после завершения терапии. Режимы дозирования для комбинированной терапии метотрексат-системные ГКС: В приведенных выше исследованиях наблюдалась значительная вариация в размере использовавшихся доз. В 2012г CARRA (Childhood Arthritis and Rheumatology Research Alliance), впервые разработали три различных варианта терапии для лечения локализованной склеродермии у детей: 1. монотерапия метотрексатом 2. метотрексат в комбинации с пульс-терапией системными ГКС с внутривенным введением метилпреднизолона 3. метотрексат и пульс терапия системными ГКС с пероральным приемом препаратов. Эти режимы дозирования были интегрированы в текущие рекомендации.
монотерапия метотрексатом 2. метотрексат в комбинации с пульс-терапией системными ГКС с внутривенным введением метилпреднизолона 3. метотрексат и пульс терапия системными ГКС с пероральным приемом препаратов. Эти режимы дозирования были интегрированы в текущие рекомендации.
Мофетила микофенолат
В 2009г мофетила микофенолат был предложен как альтернатива в случае резистентности локализованной склеродермии к метотрексату и системным ГКС. В ретроспективное исследование были включены 10 деетй с преимущественно линейной и генерализованной склеродермией. Успех терапии определялся путем инфракрасной термографии. Доза мофетила микофенолата использовалась от 600 до 1200 мг/ м² два раза в день, ежедневно, комбинация метотрексата и мофетил микофенолята использовалась в 6 случаях. Клиническое улучшение наблюдалось во всех случаях (регресс эритемы по краю очага, уменьшение склероза), что привело к значительному снижению дозы системных ГКС в пяти случаях. Несмотря на то, что данные по применению мофетила микофенолата на сегодняшний день недостаточны, он представляет собой весьма многообещающий вариант терапии второй линии по мнению авторов и CARRA..png)
Кальципотриол
В одном из немногочисленный двойных слепых плацебо-контролируемых исследований по лечению локализованной склеродермии, тераипя пероральным кальципотриолом (0,75 мкг/день в течение 6 месяцев, затем 1,25 мкг/день в течение 3 месяцев) в течение 9 месяцев не привело к значительному улучшению по сравнению с плацебо. По мнению авторов, пероральный прием кальципотриола не может рекомендоваться для лечения локализованной склеродермии.
Д-пеницилламин
Используемый в течение десятилетий для лечения системной склеродермии на сегодняшний день Д-пеницилламин признан устаревшим. Несмотря на то, что в небольших сериях клинических случаев наблюдалось улучшение на фоне приема Д-пеницилламина (2-5 мг/кг в день), контролируемых исследований не проводилось. Тем не менее, существуют клинические наблюдения в которых Д-пеницилламин применялся для лечения впервые возникшей локализованной склеродермии. С учетом спектра побочных явлений и недостаточной уверенности в эффективности препарата, авторы не могут рекомендовать Д-пеницилламин для лечения локализованной склеродермии.
Пенициллин
Так как локализованная склеродермия может возникнуть после перенесенной инфекции Borrelia spp. , в течение десятилетий для лечения локализованной склеродермии использовались пенициллин (внутривенное введение, 14-21 день) и в некоторых случаях цефтриаксон (внутривенное введение). Несмотря на то, что пенициллин обладает некоторой противовоспалительной активностью, антифибротического эффекта у препарата обнаружено не было. По мнению авторов, эффективность пенициллина не является доказанной, несмотря на наличие единичных клинических наблюдений.
Прочие препараты
Существуют клинические наблюдения, в которых отмечается успешное применение циклоспорина А, азатиоприна, хлорохина и гидроксихлорохина, фенитоина, колхицина, ретиноидов, экстракорпорального фотофереза, плазмафереза, внутривенного введения иммуноглобулинов, абатацепта, инфликсимаба, ритуксимаба, иматиниба и других вариантов лечения.
- ! Системным препаратом первого выбора при тяжелой локализованной склеродермии с вовлечением кожи и/или скелетно-мышечной системы должен быть метотрексат, с продолжительностью лечения не менее 12 месяцев. При достижении успеха от терапии возможно снижение дозы препарата.
- ! В активную фазу заболевания может применяться дополнительная терапия системными ГКС.
- ! Альтернативой метотрексату и системным ГКС в случае рефрактерности к терапии или непереносимости может служить мофетила микофенолят.
 При достижении успеха от терапии возможно снижение дозы препарата.
При достижении успеха от терапии возможно снижение дозы препарата.Физическая терапия
Исследований о роли физической терапии при локализованной склеродермии не проводилось. Тем не менее, физическая терапия является ключевым компонентом в мультимодальной схеме терапии и часто используется в повседневной клинической практике. В частности, пациенты с линейной, генерализованной, глубокой, смешанной формами локализованной склеродермии должны получать физическую терапия, за исключением острой воспалительной фазы заболевания. В фазу склероза массаж и ручной лимфодренаж должен сопровождать системную терапию либо следовать за ней. Авторы рекомендуют проводить циклы терапии один или два раза в неделю как минимум три месяца. Упражнения ЛФК и построение мышц необходимы при линейных формах локализованной склеродермии с поражением конечностей,для предотвращения образования контрактур.
- ! При подтипах локализованной склеродермии, сопровождающихся нарушением подвижности требуется физическая и мануальная терапия в качестве дополнения к топической и системной терапии. В фазу склероза массаж и лимфодренажные техники должны сопровождать или следовать за системной терапией.
Хирургическое лечение
Ортопедические/хирургическое вмешательство (например, хирургическое удлинение Ахиллова сухожилия) показаны только при линейной локализованной склеродермии. Для того, чтобы минимизировать риск обострения заболевания , оперативное вмешательство следует проводить только в неактивную фазу заболевания, в идеале – через несколько лет после исчезновения признаков активности заболевания. При подозрении на сохранение активности заболевания необходимо рассмотреть возможность инициации периоперативной системной иммуносупрессивной терапии. Эпифизиодезис здоровой ноги для коррекции разницы в длине должен производиться опытным хирургом-педиатром в период роста ребенка, например, в течение предпубертатного скачка роста. С косметической целью при подтипе en coup de sabre или прогрессирующей гемиатрофии лица может применяться аутологичная трансплантация жировой ткани, пластическая хирургия, имплантация субстанций , корректирующих косметический дефект (филлеры).
С косметической целью при подтипе en coup de sabre или прогрессирующей гемиатрофии лица может применяться аутологичная трансплантация жировой ткани, пластическая хирургия, имплантация субстанций , корректирующих косметический дефект (филлеры).
! Функционально необходимые оперативные вмешательства при линейной форме локализованной склеродермии необходим проводить в неактивную фазу заболевания.
! Пластические операции при подтипе en coup de sabre или прогрессирующей гемиатрофии лица следует проводить в неактивную фазу заболевания.
Приложение
Патогенез локализованной склеродермии
Различные формы локализованной склеродермии характеризуются хронической фиброзирующей реакцией со стороны соединительной ткани. К ранним изменениям относится образование очень плотного, первично лимфоцитарного воспалительного инфильтрата поверхностных и, в зависимости от клинического варианта, глубоких сосудов. При некоторых подтипах, например, эозинофильном фасциите, фиброз начинается с фасции. Заболевание сопровождается эозинофилией крови, а также повышенным содержанием эозинофилов, плазматических клеток и лимфоцитов в воспалительном инфильтрате, особенно в раннюю фазу заболевнаия. Считается, что при локализованной склеродермии, подобно системной склеродермии, цитокины вырабатываются воспалительным инфильтратом, затем активируют мезенхимальные клетки. Основные цитокины, вовлеченные в процесс — TFG-ß, platelet-derived growth factor (PDGF) и connective tissue growth factor (CTGF). На сегодняшний день не установлено, приводят ли эти цитокины к активации фибробластов, уже существующих в области инфильтрата или активируют дифференцировку мезенхимальных клеток-предшественников, мигрировавших в очаг.На поздних стадиях заболевания число воспальтельных клеток снижается, отмечается массивное отложение компонентов соединительной ткани, которые, при выполнении электронной микроскопии, ассоциированы с изменениями диаметра коллагеновых волокон и повышенной вариабельностью толщины фибрилл.
При некоторых подтипах, например, эозинофильном фасциите, фиброз начинается с фасции. Заболевание сопровождается эозинофилией крови, а также повышенным содержанием эозинофилов, плазматических клеток и лимфоцитов в воспалительном инфильтрате, особенно в раннюю фазу заболевнаия. Считается, что при локализованной склеродермии, подобно системной склеродермии, цитокины вырабатываются воспалительным инфильтратом, затем активируют мезенхимальные клетки. Основные цитокины, вовлеченные в процесс — TFG-ß, platelet-derived growth factor (PDGF) и connective tissue growth factor (CTGF). На сегодняшний день не установлено, приводят ли эти цитокины к активации фибробластов, уже существующих в области инфильтрата или активируют дифференцировку мезенхимальных клеток-предшественников, мигрировавших в очаг.На поздних стадиях заболевания число воспальтельных клеток снижается, отмечается массивное отложение компонентов соединительной ткани, которые, при выполнении электронной микроскопии, ассоциированы с изменениями диаметра коллагеновых волокон и повышенной вариабельностью толщины фибрилл. Перемычки между молекулами колллагена также отличаются от тех, которые характерны для нормальной дермы. Эластические волокна обычно сохранены. Гистологически обнаруживаются гомогенизированные аучки коллагеновых волокон; придатки кожи “замуровываются” и впоследствии пропадают практически бесследно по мере прогрессирования заболевания. Наличие определенного сходства между многими из этих изменений и хронической склеродермоподобно РТПХ привело к инициированию ряда исследований, посвященных дифференцировке Т-клеток и, в частности, регуляции фенотипов Th2 и Th3. Например, некоторые исследования in vitro показали, что Th3-ассоциированный цитокин интерлейкин 4 стимулирует повышение продукции коллагена фибробластами. Обнаружение этого цитокина также объясняет увеличение уровня эозинофилов на начальных стадиях заболевания. Другие исследования специализировались на молекулярных механизмах посредством которых происходит активация фибробластов. Были публикованы работы, посвященные значимости TFG-β, регуляции его активности через экспрессию рецептора TFG-β и его сигнальные пути.
Перемычки между молекулами колллагена также отличаются от тех, которые характерны для нормальной дермы. Эластические волокна обычно сохранены. Гистологически обнаруживаются гомогенизированные аучки коллагеновых волокон; придатки кожи “замуровываются” и впоследствии пропадают практически бесследно по мере прогрессирования заболевания. Наличие определенного сходства между многими из этих изменений и хронической склеродермоподобно РТПХ привело к инициированию ряда исследований, посвященных дифференцировке Т-клеток и, в частности, регуляции фенотипов Th2 и Th3. Например, некоторые исследования in vitro показали, что Th3-ассоциированный цитокин интерлейкин 4 стимулирует повышение продукции коллагена фибробластами. Обнаружение этого цитокина также объясняет увеличение уровня эозинофилов на начальных стадиях заболевания. Другие исследования специализировались на молекулярных механизмах посредством которых происходит активация фибробластов. Были публикованы работы, посвященные значимости TFG-β, регуляции его активности через экспрессию рецептора TFG-β и его сигнальные пути. Суммируя все вышесказанное, можно сказать, что существует множество причин считать, что начальная активация мезенхимальных клеток сопровождается их длительной стимуляцией через аутокринные механизмы.Несмотря на то, что процесс контроля воспалительной фазы и молекулярных механизмов, вовлеченных в развитие фиброза, изучены достаточно хорошо, об инициирующих триггерах заболевания известно совсем немного. Основываясь на клинической картине, давно появилось предположение, что инфекционный агент может играть важную роль как минимум при нескольких вариантах локализованной склеродермии. Ряд исследовательских групп рассматривали Borrelia в качестве потенциального этиологического фактора. Вероятнее всего,ь именно это является причиной столь частого назначения пенициллина в качестве начальной терапии локализованной склеродермии. Однако, в более поздних исследованиях взаимосвязь между возникновением локализованной склеродермии и присутствием Borrelia burgdorferi не была подтверждена.
Суммируя все вышесказанное, можно сказать, что существует множество причин считать, что начальная активация мезенхимальных клеток сопровождается их длительной стимуляцией через аутокринные механизмы.Несмотря на то, что процесс контроля воспалительной фазы и молекулярных механизмов, вовлеченных в развитие фиброза, изучены достаточно хорошо, об инициирующих триггерах заболевания известно совсем немного. Основываясь на клинической картине, давно появилось предположение, что инфекционный агент может играть важную роль как минимум при нескольких вариантах локализованной склеродермии. Ряд исследовательских групп рассматривали Borrelia в качестве потенциального этиологического фактора. Вероятнее всего,ь именно это является причиной столь частого назначения пенициллина в качестве начальной терапии локализованной склеродермии. Однако, в более поздних исследованиях взаимосвязь между возникновением локализованной склеродермии и присутствием Borrelia burgdorferi не была подтверждена. На сегодняшний день механизмы, лежащие в основе развития склеродермии окончательно не выяснены. Вероятнее всего, существуют различные провоцирующие факторы и также различные объекты заболевания? В совокупности с определенной генетической предрасположенностью эти факторы приводят к иммунологически опосредованной воспалительной реакции, которая сопровождается нарушением регуляции метаболизма соединительной ткани и реакции со стороны фибробластов.
На сегодняшний день механизмы, лежащие в основе развития склеродермии окончательно не выяснены. Вероятнее всего, существуют различные провоцирующие факторы и также различные объекты заболевания? В совокупности с определенной генетической предрасположенностью эти факторы приводят к иммунологически опосредованной воспалительной реакции, которая сопровождается нарушением регуляции метаболизма соединительной ткани и реакции со стороны фибробластов.
Диагностические методы оценки степени вовлечения кожи
Computerized Skin Score (CSS)
Путем оценки различных параметров Computerized Skin Score (CSS), особый компьютеризированный метод, способствует документированию распространенности активных и склерозированных/уплотненных очагов у пациентов с локализованной склеродермией. С этой целью на кожу в очаге поражения помещается Tegaderm film, затем активные и уплотненные очаги обозначаются различными цветами. Специальная компьютерная программа распознает различные обозначения и подсчитывает площадь распространения всего очага, а также размеры воспалительной границы и склерозированного центра.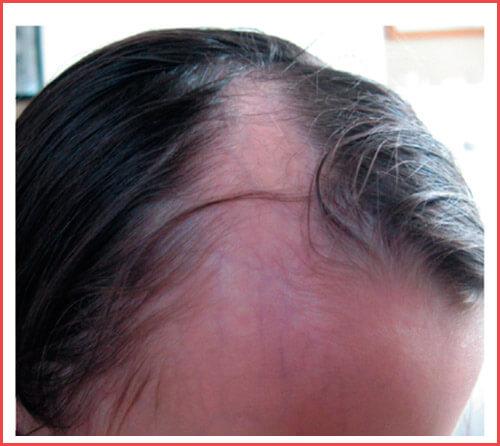 Если в программу загружены рост и вес пациента, она также рассчитывает площадь распространения очага по отношению к размерам тела пациента. Несмотря на то, что данная методика является быстрой, простой в исполнении и неинвазивной, на сегодняшний день не было проведено достаточного количества исследований, позволяющих оценить терапевтический эффект от данной техники?
Если в программу загружены рост и вес пациента, она также рассчитывает площадь распространения очага по отношению к размерам тела пациента. Несмотря на то, что данная методика является быстрой, простой в исполнении и неинвазивной, на сегодняшний день не было проведено достаточного количества исследований, позволяющих оценить терапевтический эффект от данной техники?
Ультразвук с частотой 20 МГц
Для диагностики локализованной склеродермии применялся ультразвук с частотой от 10 до 35 МГц, при этом наилучшие результаты, по данным литературы, были обнаружены при частоте 20 МГц. К примеру, при воздействии ультразвуком с более высокой частотой — около 100 МГц, наблюдается более высокая разрешающая способность, однако, глубина проникновения при такой частоте составляет лишь 1,5 мм. По сравнению с нормальной кожей, при исследовании с применением ультразвука частотой 20 МГц, обнаруживается утолщенная гипоэхогенная кожа как в воспалительную фазу локализованной склеродермии, так и в начальную фазу системной склеродермии.![]() При прогрессированнии склеротических изменений и уменьшении воспалительной инфильтрации обычно также нарастает эхогенность очага. При исследовании ювенильных форм также четко прослеживаются различия между пораженной и неизмененной кожей, а также улучшение на фоне начатой терапии. В ряде исследований подтвержден высокий уровень обоснованности, надежности и воспроизводимости результатов при обследовании пациентов с склерозирующими заболеваниями кожи с использованием ультразвукового исследования с частотой 20 МГц.
При прогрессированнии склеротических изменений и уменьшении воспалительной инфильтрации обычно также нарастает эхогенность очага. При исследовании ювенильных форм также четко прослеживаются различия между пораженной и неизмененной кожей, а также улучшение на фоне начатой терапии. В ряде исследований подтвержден высокий уровень обоснованности, надежности и воспроизводимости результатов при обследовании пациентов с склерозирующими заболеваниями кожи с использованием ультразвукового исследования с частотой 20 МГц.
Кутометр
Фиброз, возникающий при локализванной склеродермии, казывает значительное влияние на эластичность кожи. Один из методов, позволяющих оценить эластичность кожи, основывается на использовании вакуумной помпы (например, Cutometer SEM 474, Courage & Khazaka Electronic GmbH, Cologne, Germany ), расположенной перпендикулярно поверхности кожи. Деформация кожи оценивается в области тестируемой зоны(диаметр около 8 мм). Необходимо учитывать, что на эластичность кожи, оцениваемой кутометром влияют различные факторы, например, анатомическая область, в которой производится оценка, пол, возраст и другие локальные факторы, например, отечность кожи. Данная техника испльзовалась в различных исследованиях и, по мнению авторов, представляется достаточно воспроизводимой методикой оценки эластичности склерозированных участков кожи, несмотря на то, что данных литературы значительно меньше, чем в случае ультразвука частотой 20 МГц, исследований валидизирующих данный метод не проводилось.
Данная техника испльзовалась в различных исследованиях и, по мнению авторов, представляется достаточно воспроизводимой методикой оценки эластичности склерозированных участков кожи, несмотря на то, что данных литературы значительно меньше, чем в случае ультразвука частотой 20 МГц, исследований валидизирующих данный метод не проводилось.
Дурометр
В отличие от кутометра, дурометр оценивает не эластичность, а плотность кожи. Это простые в использовании инструменты, которые оценивают плотность кожи в специальных единицах при расположении специальной небольшой ручки перпендикулярно поверхности очага. В недавно опубликованном исследовании производилась оценка поражения кожи у детей с использованием дурометра, при этом были обнаружены четкие различия между пораженной и непораженной кожей, однако, для более точной оценки данного метода требуются дополнительные исследования. Несмотря на отсутствие значительных расхождений при использовании различными исследователями, четкой корреляции с другими методами оценки не было обнаружено. К тому же отсутствуют данные о чувствительности метода, особенно по отношению к обследованию пациентов после курса терапии follow-up
К тому же отсутствуют данные о чувствительности метода, особенно по отношению к обследованию пациентов после курса терапии follow-up
Термография
Воспалительные поражения подобные тем, что обнаруживаются в активную фазу локализованной склеродермии, ассоциированы с микроциркуляторными изменениями и могут опосредованно обнаруживаться при проведении термографии. Инфракрасная термография используется для измерения температуры кожи. При помощи данного метода можно оценить абсолютные значения температуры в определенной точке либо целой области, а также разницу температур. Различие в температуре пораженной и непораженной кожи позволяет судить об уровне микроциркуляции и воспалительной активности в очаге. Инфракрасная термография является валидизированным и воспроизводимым методом оценки активности заболевания при локализованной склеродермии. Чувствительность инфракрасной термографии в определении активности заболевания составляет от 0,79 до 0,92. Интересно, что частота ложно-позитивных результатов увеличивается в старых или неактивных очагах. По этой причине данный метод не рекомендуется применять в области выраженной атрофии дермы или подкожной жировой клетчатки.
По этой причине данный метод не рекомендуется применять в области выраженной атрофии дермы или подкожной жировой клетчатки.
Лазерное допплеровское исследование
При проведении лазерного допплеровского исследования кожа облучается монохроматическим лазерным лучом (длина волны 810 нм) и движение эритроцитов в сосудах тканей создает эффект Допплера. На сегодняшний день крупных проспективных работ по применению лазерного допплеровского исследования при склерозирующих заболеваниях кожи недостаточно. Слегка модифицированный метод, лазерная допплеровская визуализация (LDI) основан на применении лазерного луча(длина 690 нм), который проникает сквозь поверхность кожи. Движение крови приводит к отражению лазерного света, которое, в свою очередь, преобразуется в цветную картинку. В одном из исследований у 11 пациентов из 15 у которых при использовании LDI была диагностирована активная фаза заболевания в дальнейшем наблюдалась прогрессия заболевания, в то время как у 16 из 17 пациентов, у которых по результатам LDI была признана неактивная фаза заболевания прогрессии не наблюдалось. В другом пилотном исследовании оценивалась валидность лазерного допплеровского исследования с применением излучения с двумя длинами волны (красное и зеленое). Этот метод позволяет оценить перфузию, осуществляемую крупными сосудами (красное излучение) и кровоснабжение мелкими питающими сосудистыми веточками (питающие капилляры) (зелене излучение) на различной глубине дермы. Крупных проспективных исследований по применению лазерного допплеровскго исследования на сегодняшний день недостаточно.
В другом пилотном исследовании оценивалась валидность лазерного допплеровского исследования с применением излучения с двумя длинами волны (красное и зеленое). Этот метод позволяет оценить перфузию, осуществляемую крупными сосудами (красное излучение) и кровоснабжение мелкими питающими сосудистыми веточками (питающие капилляры) (зелене излучение) на различной глубине дермы. Крупных проспективных исследований по применению лазерного допплеровскго исследования на сегодняшний день недостаточно.
Особенности клинической картины локализованной склеродермии в детском возрасте
В то время как ограниченная локализованная склеродермия возникает преимущественно во взрослом возрасте, линейная форма локализованной склеродермии чаще встречается у детей. К возможным триггерам относят травматизацию, инфекцию, генетическую предрасположенность, нарушения эмбрионального развития. В недавних исследованиях у 65 обследованных детей было обнаружено преимущественное распространение линейной склеродермии вдоль линий Блашко, что может свидетельствовать о возможном наличии кластеров эмбриональных клеток в мозаичном состоянии. В наиболее крупном исследовании (участвовало около 750 детей) наиболее частым подтипом оказалась линейная локализованная склеродермия(с поражением конечностей) — у 65% детей, на втором месте оказалась ограниченная форма (бляшечный подтип) — у 26% обследованных, генерализованная форма — у 7% пациентов, глубокая форма — у 2 % обследованных детей. У23 % случаев пациентов обнаружено вовлечение области головы (линейная локализованная склеродермия en coup de sabre и прогрессирующая гемиатрофия лица). В 12% случаев наблюдался отягощенный семейный анамнез по ревматическим и аутоиммунным заболеваниям. Изучение 552 детей с локализованной склеродермией и системной склеродермией дало схожие результаты. Таким образом, линейная локализованная склеродермия является наиболее распространенной формой склеродермии у детей. Сочетание различных форм локализованной склеродермии не является редкостью. Как правило, ювенильная линейная локализованная склеродермия с поражением конечностей характеризуется более тяжелым течением, чем у взрослых, может приводить к значительной атрофии кожи и мышц, образованию контрактур, укорочению конечностей и уменьшению их диаметра.
В наиболее крупном исследовании (участвовало около 750 детей) наиболее частым подтипом оказалась линейная локализованная склеродермия(с поражением конечностей) — у 65% детей, на втором месте оказалась ограниченная форма (бляшечный подтип) — у 26% обследованных, генерализованная форма — у 7% пациентов, глубокая форма — у 2 % обследованных детей. У23 % случаев пациентов обнаружено вовлечение области головы (линейная локализованная склеродермия en coup de sabre и прогрессирующая гемиатрофия лица). В 12% случаев наблюдался отягощенный семейный анамнез по ревматическим и аутоиммунным заболеваниям. Изучение 552 детей с локализованной склеродермией и системной склеродермией дало схожие результаты. Таким образом, линейная локализованная склеродермия является наиболее распространенной формой склеродермии у детей. Сочетание различных форм локализованной склеродермии не является редкостью. Как правило, ювенильная линейная локализованная склеродермия с поражением конечностей характеризуется более тяжелым течением, чем у взрослых, может приводить к значительной атрофии кожи и мышц, образованию контрактур, укорочению конечностей и уменьшению их диаметра. Часто возникают выраженные функциональные, косметические и психологические нарушения. От 30 до 50% пациентов испытывают остеоартикулярные нарушения, такие как артралгии и артриты пораженной конечности. Линейная локализованная склеродермия en coup de sabre и прогрессирующая гемиатрофия лица, представляя спектр заболевания с частично пересекающимися клиническими признаками, наиболее часто возникают в раннем детском возрасте. В отличие от остальных подтипов локализованной склеродермии, они имеют более медленное прогрессирующее течение, часто характеризующееся длительной активной фазой. Часто сопровождаются неврологическими симптомами, такими как эпилептические припадки, нейропсихиатрические симптомы, головная боль, нарушение поведения, снижение способности к обучаемости. При выполнении биопсии головного мозга обнаруживается глиоз, периваскулярный воспалительный инфильтрат. В зависимости от тяжести и распространенности процесса поражение может охватывать щеки, нос, верхную губу, подбородок.
Часто возникают выраженные функциональные, косметические и психологические нарушения. От 30 до 50% пациентов испытывают остеоартикулярные нарушения, такие как артралгии и артриты пораженной конечности. Линейная локализованная склеродермия en coup de sabre и прогрессирующая гемиатрофия лица, представляя спектр заболевания с частично пересекающимися клиническими признаками, наиболее часто возникают в раннем детском возрасте. В отличие от остальных подтипов локализованной склеродермии, они имеют более медленное прогрессирующее течение, часто характеризующееся длительной активной фазой. Часто сопровождаются неврологическими симптомами, такими как эпилептические припадки, нейропсихиатрические симптомы, головная боль, нарушение поведения, снижение способности к обучаемости. При выполнении биопсии головного мозга обнаруживается глиоз, периваскулярный воспалительный инфильтрат. В зависимости от тяжести и распространенности процесса поражение может охватывать щеки, нос, верхную губу, подбородок. Возможны различные офтальмологические нарушения — увеит, дисфункция глазодвигательных мышц, потеря бровей, деформация век. Достаточно редкая генерализованная форма — пансклеротическая морфеа обычно возникает впервые до 14 лет и характеризуется быстрым прогрессирующим течением с обязательным вовлечением в процесс внекожных структур (подкожной жировой клетчатки, мышц, костей). Часто сопровождается тяжелым замедлением роста, кахексией. Клиническое течение бляшечной формы, глубокой склеродермии и эозинофильного фасциита не отличается от взрослых пациентов, также может сочетаться с другими формами локализованной склеродермии.
Возможны различные офтальмологические нарушения — увеит, дисфункция глазодвигательных мышц, потеря бровей, деформация век. Достаточно редкая генерализованная форма — пансклеротическая морфеа обычно возникает впервые до 14 лет и характеризуется быстрым прогрессирующим течением с обязательным вовлечением в процесс внекожных структур (подкожной жировой клетчатки, мышц, костей). Часто сопровождается тяжелым замедлением роста, кахексией. Клиническое течение бляшечной формы, глубокой склеродермии и эозинофильного фасциита не отличается от взрослых пациентов, также может сочетаться с другими формами локализованной склеродермии.
Особые серологические и диагностические признаки локализованной склеродермии у детей
Как было сказано ранее, детская локализованная склеродермия часто сопровождается серологическими изменениями. В активную фазу генерализованной формы может наблюдаться эозинофилия. Лабораторные изменения чаще всего обнаруживаются при линейной форме. В активную фазу заболевания может обнаруживаться повышение ревматоидного фактора, СОЭ, гипергаммаглобулинемия ( повышение IgA и IgM в активную фазу заболевания и повышение IgG при тяжелой форме с образованием контрактур). Антинуклеарные антитела часто характеризуются гомогенностью. Повышение анти-гистоновых и анти- ssDNA антител может наблюдаться при распространенных линейных формах с вовлечением суставов. Антитела против экстрагируемых ядерных антигенов часто не определяются. С учетом частого вовлечения суставов при линейной форме локализованной склеродермии, при необходимости следует выполнять инструментальное обследование суставов (УЗИ, рентген, МРТ, сцинтиграфию костей). При наличии неврологических или офтальмологических симптомов, ассоциированных с линейной формой локализованной склеродермии en coup de sabre или прогрессирующей гемиатрофией лица необходимо выполнять МРТ. Несмотря на то, что в межнациональном исследовании 750 пациентов с ювенильной локализованной склеродермией было обнаружено системное вовлечение (легкие — 2,6%, сердце — 1%, почки — 1%) авторы рекомендаций не наблюдали подобных изменений, поэтому , по мнению авторов, рутинное обследование для исключения системного процесса при ювенильной локализованной склеродермии не рекомендуется.
Антинуклеарные антитела часто характеризуются гомогенностью. Повышение анти-гистоновых и анти- ssDNA антител может наблюдаться при распространенных линейных формах с вовлечением суставов. Антитела против экстрагируемых ядерных антигенов часто не определяются. С учетом частого вовлечения суставов при линейной форме локализованной склеродермии, при необходимости следует выполнять инструментальное обследование суставов (УЗИ, рентген, МРТ, сцинтиграфию костей). При наличии неврологических или офтальмологических симптомов, ассоциированных с линейной формой локализованной склеродермии en coup de sabre или прогрессирующей гемиатрофией лица необходимо выполнять МРТ. Несмотря на то, что в межнациональном исследовании 750 пациентов с ювенильной локализованной склеродермией было обнаружено системное вовлечение (легкие — 2,6%, сердце — 1%, почки — 1%) авторы рекомендаций не наблюдали подобных изменений, поэтому , по мнению авторов, рутинное обследование для исключения системного процесса при ювенильной локализованной склеродермии не рекомендуется.
Особенности лечения локализованной склеродермии в детском возрасте
Для того, чтобы избежать последующих осложнений (контрактур и деформации конечностей), по мнению авторов, необходимо раннее начало интенсивной системной терапии у детей с линейной формой локализованной склеродермии. Как и у взрослых, выбор терапии должен основываться на подтипе заболевания и локализации поражения. Необходима сопутствующая физиотерапия, включая ручной лимфодренаж. Ортопедические процедуры (например, хирургические процедуры — удлинение Ахиллова сухожилия, эпифизиодез здоровой нижней конечности для коррекции разницы в длине ) необходимо проводить в длительно протекащую неактивную фазу заболевания. Это касается и проведения косметических процедур при линейной локализованной склеродермии en coup de sabre и прогрессирующей гемиатрофии лица.
Склеродермия — ПроМедицина Уфа
Склеродерма (склеродермия)
— диффузное заболевание соединительной ткани, при котором в отдельных участках кожи, а иногда и во всей коже, откладывается рубцовая ткань. При распространении процесса рубцовая ткань образуется не только в коже, но и в опорно-двигательном аппарате, легких, пищеварительном тракте, сердце и почках.
При распространении процесса рубцовая ткань образуется не только в коже, но и в опорно-двигательном аппарате, легких, пищеварительном тракте, сердце и почках.Склеродермия является аутоиммунным ревматологическим заболеванием. Термин «аутоиммунный» означает, что в основе данного недуга лежит нарушение работы иммунной системы, которая по каким-либо причинам начинает «атаковать» клетки и ткани собственного организма. Следствием этого является воспалительная реакция, которая ведет к истончению и ужесточению кожных покровов, кровеносных сосудов, а также ряда внутренних органов (таких как пищевод, легкие, почки, желудок, кишечник, сердце).
Причины
Причины заболевания неизвестны. Считается, что склеродермия развивается под влиянием некоторых внешних факторов у людей с определенными генетическими нарушениями. К внешним факторам, способным провоцировать развитие склеродермии, относятся
— ретровирусы,
— кварцевая и каменноугольная пыль,
— органические растворители,
— винилхлорид,
— некоторые лекарственные средства (блеомицин и ряд других препаратов, применяемых для химиотерапии).
Симптомы
Склеродермия проявляется различными клиническими симптомами, которые зависят от локализации процесса и, соответственно, от пораженных органов. Немаловажным фактором, влияющим на проявления патологии, является стадия ее развития и степень склеротических изменений.
На начальных стадиях склеродермии могут возникать следующие неспецифические симптомы:
— недомогание;
— усталость;
— боль в суставах;
— боль в мышцах.
У некоторых пациентов может наблюдаться одышка и изжога (указывают на поражение внутренних органов).
Дальнейшая симптоматика склеродермии зависит от локализации и распространения склеротических очагов.
При ограниченной склеродермии возникают отдельные очаги в толще кожи, иногда – мышц и костей. Развивается поражение периферических кровеносных сосудов. В зависимости от формы и типа очагов различают бляшечную, линейную и пятнистую склеродермию.
При системной склеродермии развивающиеся очаги не ограничиваются поражением кожи и распространяются во внутренние органы, вызывая нарушение их функции.
Так как в основе склеродермии лежит замещение нормальной ткани на анормальные соединительнотканные волокна, в пораженных областях и органах наблюдается значительное снижение эластичности, растяжимости и подвижности. В результате органы и конечности не могут адекватно реагировать на стимулы, что и формирует основу клинической картины.
Диагностика
Диагностика склеродермии затруднена, так как симптоматика часто схожа с массой других заболеваний, эта аномалия не имеет собственных характерных особенностей. Именно поэтому сначала исключают схожие диагнозы, оценивается наличие главных признаков склеродермии:
— Поражения кожных покровов, в том числе появление бляшек и участков, отличных по структуре и цвету от здоровых участков кожи.
— Изменения функций мышц и суставов, от незначительных отклонений до полной атрофии
— Положительный синдром Рейно, который проявляется некрозом верхних слоёв эпителия пальцев конечностей.
— Нарушение кровоснабжения на разных участках тела и в разных органах.
— Отклонения в работе жизненно важных систем, в первую очередь со стороны желудочно-кишечного тракта, но при условии отсутствия хронических заболеваний в этих органах.
Если во время опроса пациента, врачебного осмотра, разных методов компьютерной диагностики и лабораторных исследований были выявлены три и более основных признака, то может быть поставлен первичный диагноз.
Лечение
Возможности медицины в борьбе с дерматосклерозом ограничены. Основным направлением в лечении склеродермии является устранение или облегчение симптомов. Разрабатывается индивидуальная программа комплекса упражнений лечебной физкультуры для развития суставной моторики. Для этой же цели назначается лечебный массаж и различные методики трудотерапии.
Рекомендованы: грязевые и парафиновые аппликации; на пораженные конечности – процедуры электрофореза и плазмафереза; диадинамические токи и ультразвук.
В качестве медикаментозной терапии назначаются: препараты, которые подавляют фиброобразование на соединительных тканях; ферменты, которые расщепляют уже образованные уплотнения; противовоспалительные и общеукрепляющие препараты; витаминотерапию и вазоактивную терапию; антибиотики. Самолечение при таком серьезном заболевании неуместно.
Самолечение при таком серьезном заболевании неуместно.
Педиатрическая системная склеродермия
Обзор
Что такое склеродермия?
Склеродермия — это состояние, при котором кожа становится необычно толстой и твердой. Это аутоиммунное заболевание, вызывающее воспаление (отек) кожи. Отек может вызвать перепроизводство клеток коллагена, волокнистого белка, который является основной частью многих тканей организма. Это перепроизводство может привести к фиброзу или рубцеванию.
Какие бывают виды склеродермии?
Существует два основных типа склеродермии:
- Локализованная склеродермия : Эта форма склеродермии поражает кожу только на некоторых участках тела и лишь иногда распространяется на подлежащие мышцы или кости.Она редко поражает внутренние органы и не переходит в генерализованные формы склеродермии.
- Системная склеродермия (системный склероз) : Эта форма склеродермии может вызвать генерализованное утолщение кожи во всех частях тела. Помимо кожных изменений, рубцовая ткань может развиваться во внутренних органах, таких как почки, сердце, легкие и желудочно-кишечный тракт. У детей с системным склерозом более распространены кожные изменения, которые могут привести к ограничению движений в суставах.
 Помимо кожных изменений, рубцовая ткань может развиваться во внутренних органах, таких как почки, сердце, легкие и желудочно-кишечный тракт. У детей с системным склерозом более распространены кожные изменения, которые могут привести к ограничению движений в суставах.
Помимо кожных изменений, рубцовая ткань может развиваться во внутренних органах, таких как почки, сердце, легкие и желудочно-кишечный тракт. У детей с системным склерозом более распространены кожные изменения, которые могут привести к ограничению движений в суставах.Феномен Рейно (изменение белого, синего и красного цвета пальцев рук и/или ног при воздействии холода или стресса) присутствует в раннем возрасте у ребенка с системным склерозом, а также утомляемость, боль в суставах, затрудненное глотание, боль в животе, изжога, диарея и одышка.
Детей с системной склеродермией следует часто проверять на наличие высокого кровяного давления, а также проблем с легкими, почками, желудочно-кишечным трактом и сердцем.
Кто болеет склеродермией?
В США всего от 5000 до 7000 детей со склеродермией.Чаще встречается у взрослых, чем у детей. Только у двух процентов всех людей со склеродермией заболевание развивается в возрасте до 10 лет, а у семи процентов оно развивается в возрасте от 10 до 19 лет.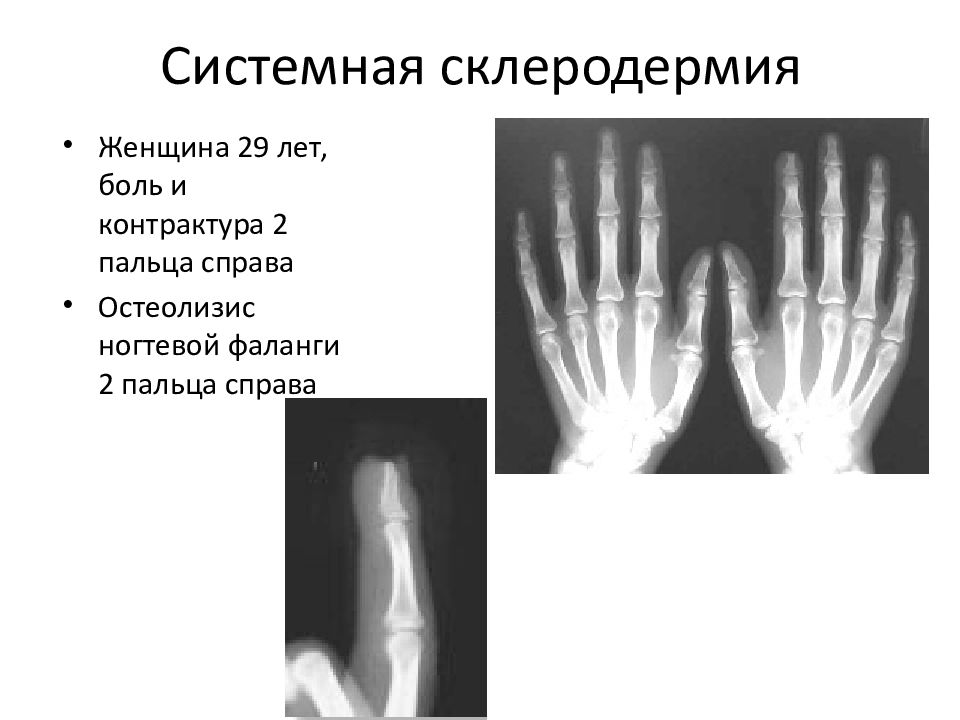
Симптомы и причины
Что вызывает системную склеродермию у детей?
Точная причина детской системной склеродермии неизвестна. По-видимому, это связано с нарушением клеток, выстилающих мелкие кровеносные сосуды.
Это также может быть связано с дефектами иммунной системы, при которых обычно защитная система организма вызывает повреждение собственных тканей.
В норме иммунная система помогает защитить организм от инфекции. У пациентов со склеродермией иммунная система заставляет другие клетки вырабатывать слишком много коллагена (белка). Этот дополнительный коллаген откладывается в коже и органах, что вызывает затвердевание и утолщение (аналогично процессу рубцевания).
Каковы признаки и симптомы детской системной склеродермии?
Изменения кожи при системной склеродермии могут включать:
- Потеря способности кожи растягиваться
- Снижение функции кисти (из-за натяжения кожи на пальцах и кисти)
- Феномен Рейно (аномальная чувствительность к холоду), который обычно наблюдается на руках. Признаки включают изменение цвета рук (белый, синий, красный), покалывание, дискомфорт и снижение чувствительности.
- Расширенные красные кровеносные сосуды на руках, лице и вокруг ногтевых лож (телеангиэктазии)
- Отложения кальция на коже или других участках (кальциноз)
 Признаки включают изменение цвета рук (белый, синий, красный), покалывание, дискомфорт и снижение чувствительности.
Признаки включают изменение цвета рук (белый, синий, красный), покалывание, дискомфорт и снижение чувствительности.Сначала кожа рук и ног кажется отечной. Со временем кожа стягивается и твердеет, на ней могут появиться гребни, углубления или небольшие ямки, которые видны в основном на кончиках пальцев.
Поскольку боли не так много, склеродермия обычно присутствует в течение некоторого времени, прежде чем родитель или ребенок забеспокоится.
Другие признаки и симптомы системной склеродермии из-за поражения внутренних органов могут включать:
- Воспаление суставов с тугоподвижностью и болью
- Язвы (язвы), в основном на кончиках пальцев
- Проблемы с пищеварением (изжога, затрудненное глотание, диарея, желудочные спазмы)
- Респираторные проблемы (хронический кашель, затрудненное дыхание, утомляемость)
- Поражение почек может привести к артериальной гипертензии
- Усталость (легко устает)
- Мышечная слабость
Диагностика и тесты
Как диагностируется системная склеродермия у детей?
Диагноз системной склеродермии часто ставится ревматологом (специалист по артритам/аутоиммунным заболеваниям) или дерматологом (специалист по кожным заболеваниям) на основании комбинации факторов, в том числе:
- Наличие симптомов и физических признаков
- Характерные изменения кожи (включая локализацию, размер, форму, цвет)
- Результаты полного медицинского анамнеза и медицинского осмотра
Специфических тестов для диагностики системной склеродермии не существует. Лабораторные тесты используются для исключения других подобных заболеваний, для оценки активности склеродермии и для определения того, поражены ли другие органы, кроме кожи. Тестирование на некоторые аутоиммунные белки может помочь определить течение заболевания. Биопсия кожи обычно не проводится.
Лабораторные тесты используются для исключения других подобных заболеваний, для оценки активности склеродермии и для определения того, поражены ли другие органы, кроме кожи. Тестирование на некоторые аутоиммунные белки может помочь определить течение заболевания. Биопсия кожи обычно не проводится.
Дополнительные тесты на системную склеродермию включают:
- Анализы крови, в которых измеряются аутоиммунные белки и функция почек
- Рентгеновские снимки, которые могут показать изменения кожи, костей и внутренних органов (таких как легкие и кишечник)
- Тесты для оценки глотательной функции пищевода (трубки, идущей изо рта в желудок)
- Проверка функции легких (дыхательная проба), чтобы убедиться, что легкие работают правильно
- Эхокардиография (УЗИ сердца) для проверки работы сердца
Управление и лечение
Как лечится детская склеродермия?
От склеродермии нет лекарства, но при правильной диагностике ее можно лечить и контролировать.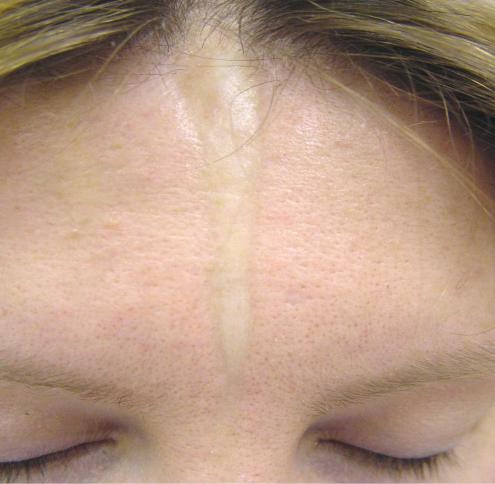 Целью лечения является остановить воспаление, предотвратить ухудшение заболевания и предотвратить поражение внутренних органов.
Целью лечения является остановить воспаление, предотвратить ухудшение заболевания и предотвратить поражение внутренних органов.
Выбор наиболее подходящего лечения осуществляется детским ревматологом, имеющим опыт лечения склеродермии, совместно с другими специалистами, занимающимися конкретными системами (желудочно-кишечный тракт, сердце, легкие и почки).
Лекарства- Кортикостероиды могут использоваться для уменьшения воспаления в мышцах, суставах или, реже, в самой коже.Стероиды также могут быть полезны при лечении ранних стадий воспаления внутренних органов. Как правило, стероиды не действуют на поздних стадиях системного склероза.
- Нестероидные противовоспалительные препараты (НПВП), такие как ибупрофен и напроксен, иногда назначают детям с артритом для уменьшения воспаления суставов.
- Лекарства, расширяющие кровеносные сосуды и улучшающие кровоток, часто используются для лечения феномена Рейно.
Другие методы лечения в настоящее время изучаются, и есть конкретная надежда, что в будущем будут найдены более эффективные методы лечения. В очень тяжелых случаях может быть рассмотрена аутологичная трансплантация костного мозга.
В очень тяжелых случаях может быть рассмотрена аутологичная трансплантация костного мозга.
Защита кожи поможет максимизировать приток крови к коже, рукам и ногам, особенно у детей с синдромом Рейно. Вот несколько советов:
- Избегайте травм пораженных участков, особенно кончиков пальцев рук и ног.
- Защищайте руки и ноги ребенка от холода. Поддерживайте в комнатах теплую температуру и надевайте на ребенка дополнительный слой одежды зимой, а также шапку с наушниками, перчатки и теплые носки.Шерсть теплее, чем хлопок или синтетические ткани, а несколько слоев тонкой одежды лучше, чем один тяжелый или толстый слой одежды.
- Избегайте курения или воздействия дыма.
- Избегайте лекарств от простуды, содержащих псевдоэфедрин.
- Защитите ребенка от чрезмерного пребывания на солнце и используйте солнцезащитные кремы в соответствии с рекомендациями.
- Избегайте использования на коже вяжущих средств, скрабов для тела или лица или агрессивных моющих средств.
- Используйте лосьоны, назначенные врачом, чтобы кожа вашего ребенка оставалась мягкой.

Базовые программы упражнений на растяжку и упражнения под руководством физиотерапевта и эрготерапевта помогают ребенку сохранить гибкость, диапазон движений в суставах, мышечную силу и приток крови к пораженным участкам. Терапия также поможет предотвратить контрактуры суставов (изгибы в суставах). При необходимости могут быть рекомендованы шины.
ХирургияВ редких случаях может потребоваться ортопедическая хирургия кисти или косметическая хирургия для коррекции тяжелых контрактур суставов, деформаций кожи или рубцов.Перед проведением хирургического вмешательства болезнь обычно должна находиться в стадии ремиссии (не активной) в течение нескольких лет.
Профилактика
Можно ли предотвратить системную склеродермию у детей?
Не существует известного способа предотвращения системной склеродермии. Это не заразно: вы не можете заразиться склеродермией при рукопожатии, совместном использовании столовых приборов, объятиях, поцелуях, сексуальном контакте и контакте с кровью или биологическими жидкостями, а также воздушно-капельным путем при кашле или чихании.
Это не заразно: вы не можете заразиться склеродермией при рукопожатии, совместном использовании столовых приборов, объятиях, поцелуях, сексуальном контакте и контакте с кровью или биологическими жидкостями, а также воздушно-капельным путем при кашле или чихании.
Перспективы/прогноз
Каков прогноз (перспективы) для детей с системной склеродермией у детей?
Системная склеродермия — хроническое (длительное) и медленно развивающееся заболевание, длящееся месяцы или годы.Прогноз зависит от того, насколько поражена кожа и затронуты ли внутренние органы.
Дети с заболеваниями легких, сердца или почек подвергаются наибольшему риску осложнений, включая контрактуры суставов, косметические изменения и неравномерный рост костей.
Хотя системная склеродермия часто не проходит, она может оставаться на том же уровне без ухудшения в течение нескольких лет.
Жить с
Что беспокоит детей с системной сцеродермией?
Дети с системной склеродермией должны жить как можно более нормальной жизнью.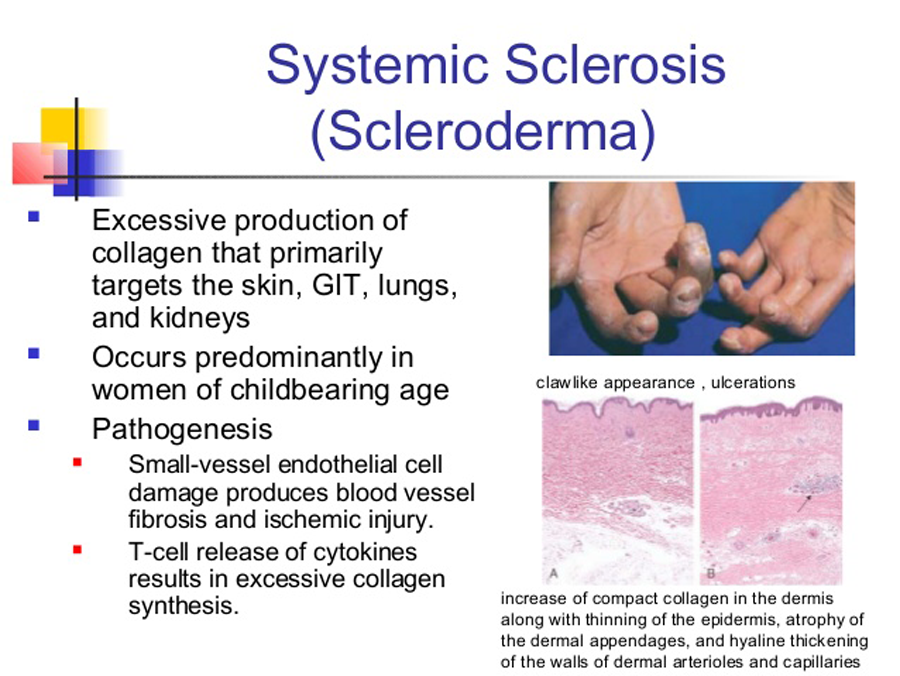 Они должны посещать школу, заниматься спортом, участвовать во внеклассных и семейных мероприятиях. В целом, нет ограничений на физическую активность детей (при условии, что она безопасна). Упражнения помогут предотвратить ухудшение состояния, повысить мышечную силу и мышечную выносливость.
Они должны посещать школу, заниматься спортом, участвовать во внеклассных и семейных мероприятиях. В целом, нет ограничений на физическую активность детей (при условии, что она безопасна). Упражнения помогут предотвратить ухудшение состояния, повысить мышечную силу и мышечную выносливость.
Не существует специальной диеты, которая доказала бы свою эффективность при склеродермии. Дети со склеродермией должны соблюдать здоровую сбалансированную диету.
Ресурсы
Существуют ли группы поддержки системной склеродермии?
Фонд склеродермии — ведущая некоммерческая организация, деятельность которой направлена на удовлетворение потребностей взрослых и детей, живущих со склеродермией в Соединенных Штатах.Многие местные и национальные мероприятия организуются для обучения и объединения пациентов и их семей.
Дополнительную информацию можно найти на сайте www.scleroderma.org.
Педиатрическая системная склеродермия
Обзор
Что такое склеродермия?
Склеродермия — это состояние, при котором кожа становится необычно толстой и твердой. Это аутоиммунное заболевание, вызывающее воспаление (отек) кожи. Отек может вызвать перепроизводство клеток коллагена, волокнистого белка, который является основной частью многих тканей организма.Это перепроизводство может привести к фиброзу или рубцеванию.
Это аутоиммунное заболевание, вызывающее воспаление (отек) кожи. Отек может вызвать перепроизводство клеток коллагена, волокнистого белка, который является основной частью многих тканей организма.Это перепроизводство может привести к фиброзу или рубцеванию.
Какие бывают виды склеродермии?
Существует два основных типа склеродермии:
- Локализованная склеродермия : Эта форма склеродермии поражает кожу только на некоторых участках тела и лишь иногда распространяется на подлежащие мышцы или кости. Она редко поражает внутренние органы и не переходит в генерализованные формы склеродермии.
- Системная склеродермия (системный склероз) : Эта форма склеродермии может вызвать генерализованное утолщение кожи во всех частях тела.Помимо кожных изменений, рубцовая ткань может развиваться во внутренних органах, таких как почки, сердце, легкие и желудочно-кишечный тракт. У детей с системным склерозом более распространены кожные изменения, которые могут привести к ограничению движений в суставах.

Феномен Рейно (изменение белого, синего и красного цвета пальцев рук и/или ног при воздействии холода или стресса) присутствует в раннем возрасте у ребенка с системным склерозом, а также утомляемость, боль в суставах, затрудненное глотание, боль в животе, изжога, диарея и одышка.
Детей с системной склеродермией следует часто проверять на наличие высокого кровяного давления, а также проблем с легкими, почками, желудочно-кишечным трактом и сердцем.
Кто болеет склеродермией?
В США всего от 5000 до 7000 детей со склеродермией. Чаще встречается у взрослых, чем у детей. Только у двух процентов всех людей со склеродермией заболевание развивается в возрасте до 10 лет, а у семи процентов оно развивается в возрасте от 10 до 19 лет.
Симптомы и причины
Что вызывает системную склеродермию у детей?
Точная причина детской системной склеродермии неизвестна.По-видимому, это связано с нарушением клеток, выстилающих мелкие кровеносные сосуды.
Это также может быть связано с дефектами иммунной системы, при которых обычно защитная система организма вызывает повреждение собственных тканей.
В норме иммунная система помогает защитить организм от инфекции. У пациентов со склеродермией иммунная система заставляет другие клетки вырабатывать слишком много коллагена (белка). Этот дополнительный коллаген откладывается в коже и органах, что вызывает затвердевание и утолщение (аналогично процессу рубцевания).
Каковы признаки и симптомы детской системной склеродермии?
Изменения кожи при системной склеродермии могут включать:
- Потеря способности кожи растягиваться
- Снижение функции кисти (из-за натяжения кожи на пальцах и кисти)
- Феномен Рейно (аномальная чувствительность к холоду), который обычно наблюдается на руках. Признаки включают изменение цвета рук (белый, синий, красный), покалывание, дискомфорт и снижение чувствительности.
- Расширенные красные кровеносные сосуды на руках, лице и вокруг ногтевых лож (телеангиэктазии)
- Отложения кальция на коже или других участках (кальциноз)
Сначала кожа рук и ног кажется отечной. Со временем кожа стягивается и твердеет, на ней могут появиться гребни, углубления или небольшие ямки, которые видны в основном на кончиках пальцев.
Со временем кожа стягивается и твердеет, на ней могут появиться гребни, углубления или небольшие ямки, которые видны в основном на кончиках пальцев.
Поскольку боли не так много, склеродермия обычно присутствует в течение некоторого времени, прежде чем родитель или ребенок забеспокоится.
Другие признаки и симптомы системной склеродермии из-за поражения внутренних органов могут включать:
- Воспаление суставов с тугоподвижностью и болью
- Язвы (язвы), в основном на кончиках пальцев
- Проблемы с пищеварением (изжога, затрудненное глотание, диарея, желудочные спазмы)
- Респираторные проблемы (хронический кашель, затрудненное дыхание, утомляемость)
- Поражение почек может привести к артериальной гипертензии
- Усталость (легко устает)
- Мышечная слабость
Диагностика и тесты
Как диагностируется системная склеродермия у детей?
Диагноз системной склеродермии часто ставится ревматологом (специалист по артритам/аутоиммунным заболеваниям) или дерматологом (специалист по кожным заболеваниям) на основании комбинации факторов, в том числе:
- Наличие симптомов и физических признаков
- Характерные изменения кожи (включая локализацию, размер, форму, цвет)
- Результаты полного медицинского анамнеза и медицинского осмотра
Специфических тестов для диагностики системной склеродермии не существует.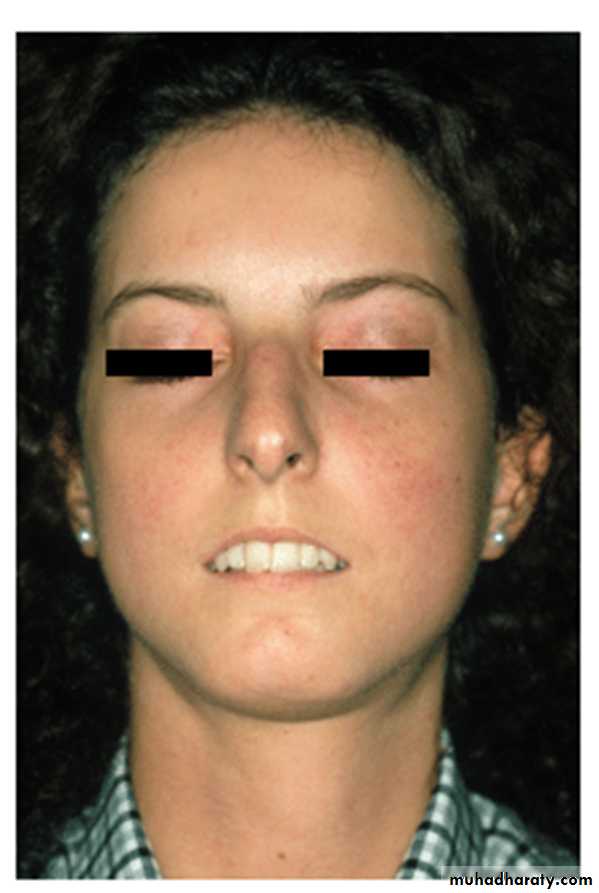 Лабораторные тесты используются для исключения других подобных заболеваний, для оценки активности склеродермии и для определения того, поражены ли другие органы, кроме кожи. Тестирование на некоторые аутоиммунные белки может помочь определить течение заболевания. Биопсия кожи обычно не проводится.
Лабораторные тесты используются для исключения других подобных заболеваний, для оценки активности склеродермии и для определения того, поражены ли другие органы, кроме кожи. Тестирование на некоторые аутоиммунные белки может помочь определить течение заболевания. Биопсия кожи обычно не проводится.
Дополнительные тесты на системную склеродермию включают:
- Анализы крови, в которых измеряются аутоиммунные белки и функция почек
- Рентгеновские снимки, которые могут показать изменения кожи, костей и внутренних органов (таких как легкие и кишечник)
- Тесты для оценки глотательной функции пищевода (трубки, идущей изо рта в желудок)
- Проверка функции легких (дыхательная проба), чтобы убедиться, что легкие работают правильно
- Эхокардиография (УЗИ сердца) для проверки работы сердца
Управление и лечение
Как лечится детская склеродермия?
От склеродермии нет лекарства, но при правильной диагностике ее можно лечить и контролировать.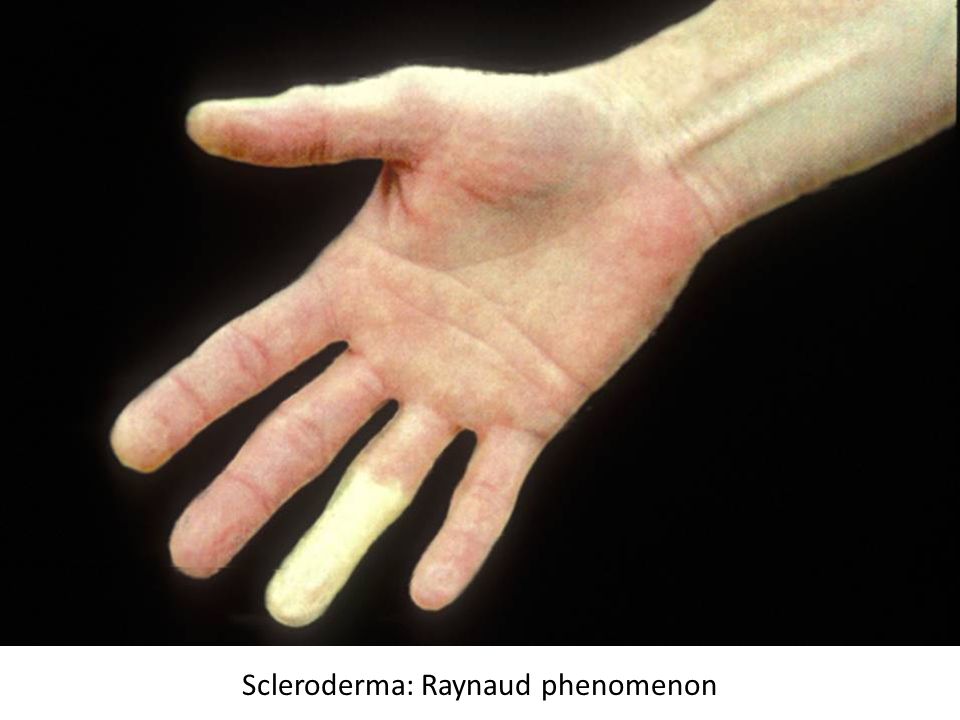 Целью лечения является остановить воспаление, предотвратить ухудшение заболевания и предотвратить поражение внутренних органов.
Целью лечения является остановить воспаление, предотвратить ухудшение заболевания и предотвратить поражение внутренних органов.
Выбор наиболее подходящего лечения осуществляется детским ревматологом, имеющим опыт лечения склеродермии, совместно с другими специалистами, занимающимися конкретными системами (желудочно-кишечный тракт, сердце, легкие и почки).
Лекарства- Кортикостероиды могут использоваться для уменьшения воспаления в мышцах, суставах или, реже, в самой коже.Стероиды также могут быть полезны при лечении ранних стадий воспаления внутренних органов. Как правило, стероиды не действуют на поздних стадиях системного склероза.
- Нестероидные противовоспалительные препараты (НПВП), такие как ибупрофен и напроксен, иногда назначают детям с артритом для уменьшения воспаления суставов.
- Лекарства, расширяющие кровеносные сосуды и улучшающие кровоток, часто используются для лечения феномена Рейно.
Другие методы лечения в настоящее время изучаются, и есть конкретная надежда, что в будущем будут найдены более эффективные методы лечения. В очень тяжелых случаях может быть рассмотрена аутологичная трансплантация костного мозга.
В очень тяжелых случаях может быть рассмотрена аутологичная трансплантация костного мозга.
Защита кожи поможет максимизировать приток крови к коже, рукам и ногам, особенно у детей с синдромом Рейно. Вот несколько советов:
- Избегайте травм пораженных участков, особенно кончиков пальцев рук и ног.
- Защищайте руки и ноги ребенка от холода. Поддерживайте в комнатах теплую температуру и надевайте на ребенка дополнительный слой одежды зимой, а также шапку с наушниками, перчатки и теплые носки.Шерсть теплее, чем хлопок или синтетические ткани, а несколько слоев тонкой одежды лучше, чем один тяжелый или толстый слой одежды.
- Избегайте курения или воздействия дыма.
- Избегайте лекарств от простуды, содержащих псевдоэфедрин.
- Защитите ребенка от чрезмерного пребывания на солнце и используйте солнцезащитные кремы в соответствии с рекомендациями.
- Избегайте использования на коже вяжущих средств, скрабов для тела или лица или агрессивных моющих средств.
- Используйте лосьоны, назначенные врачом, чтобы кожа вашего ребенка оставалась мягкой.

Базовые программы упражнений на растяжку и упражнения под руководством физиотерапевта и эрготерапевта помогают ребенку сохранить гибкость, диапазон движений в суставах, мышечную силу и приток крови к пораженным участкам. Терапия также поможет предотвратить контрактуры суставов (изгибы в суставах). При необходимости могут быть рекомендованы шины.
ХирургияВ редких случаях может потребоваться ортопедическая хирургия кисти или косметическая хирургия для коррекции тяжелых контрактур суставов, деформаций кожи или рубцов.Перед проведением хирургического вмешательства болезнь обычно должна находиться в стадии ремиссии (не активной) в течение нескольких лет.
Профилактика
Можно ли предотвратить системную склеродермию у детей?
Не существует известного способа предотвращения системной склеродермии. Это не заразно: вы не можете заразиться склеродермией при рукопожатии, совместном использовании столовых приборов, объятиях, поцелуях, сексуальном контакте и контакте с кровью или биологическими жидкостями, а также воздушно-капельным путем при кашле или чихании.
Это не заразно: вы не можете заразиться склеродермией при рукопожатии, совместном использовании столовых приборов, объятиях, поцелуях, сексуальном контакте и контакте с кровью или биологическими жидкостями, а также воздушно-капельным путем при кашле или чихании.
Перспективы/прогноз
Каков прогноз (перспективы) для детей с системной склеродермией у детей?
Системная склеродермия — хроническое (длительное) и медленно развивающееся заболевание, длящееся месяцы или годы.Прогноз зависит от того, насколько поражена кожа и затронуты ли внутренние органы.
Дети с заболеваниями легких, сердца или почек подвергаются наибольшему риску осложнений, включая контрактуры суставов, косметические изменения и неравномерный рост костей.
Хотя системная склеродермия часто не проходит, она может оставаться на том же уровне без ухудшения в течение нескольких лет.
Жить с
Что беспокоит детей с системной сцеродермией?
Дети с системной склеродермией должны жить как можно более нормальной жизнью. Они должны посещать школу, заниматься спортом, участвовать во внеклассных и семейных мероприятиях. В целом, нет ограничений на физическую активность детей (при условии, что она безопасна). Упражнения помогут предотвратить ухудшение состояния, повысить мышечную силу и мышечную выносливость.
Они должны посещать школу, заниматься спортом, участвовать во внеклассных и семейных мероприятиях. В целом, нет ограничений на физическую активность детей (при условии, что она безопасна). Упражнения помогут предотвратить ухудшение состояния, повысить мышечную силу и мышечную выносливость.
Не существует специальной диеты, которая доказала бы свою эффективность при склеродермии. Дети со склеродермией должны соблюдать здоровую сбалансированную диету.
Ресурсы
Существуют ли группы поддержки системной склеродермии?
Фонд склеродермии — ведущая некоммерческая организация, деятельность которой направлена на удовлетворение потребностей взрослых и детей, живущих со склеродермией в Соединенных Штатах.Многие местные и национальные мероприятия организуются для обучения и объединения пациентов и их семей.
Дополнительную информацию можно найти на сайте www.scleroderma.org.
Детская склеродермия – системные и локализованные формы
Краткое изложение
Детская склеродермия включает две основные группы клинических форм: системный склероз (СС) и локализованную склеродермию (ЛС). Хотя оба имеют общую патофизиологию с начальной воспалительной фазой, связанной с эндотелиальной активацией, и более поздней фиброзной фазой, проявляющейся коллагенизацией ткани и заметной толщиной кожи, их клинические проявления различаются.СЛ, как правило, ограничивается кожей и подкожным слоем, и хотя он не является смертельным, как ССД, до четверти пациентов могут иметь внекожные проявления заболевания, такие как артрит и увеит. Хотя при СС может поражаться любой орган, у детей чаще всего наблюдаются поражения сосудов (феномен Рейно), кожи (утолщение кожи), желудочно-кишечного тракта, легких и опорно-двигательного аппарата. Профили аутоантител при ССД в детском возрасте могут помочь в прогнозировании поражения внутренних органов. Лечение обеих форм склеродермии нацелено на активную воспалительную стадию и останавливает прогрессирование заболевания; тем не менее, все еще необходимо добиться прогресса в разработке более эффективной антифиброзной терапии, чтобы помочь обратить вспять повреждение, вызванное болезнью.
Хотя оба имеют общую патофизиологию с начальной воспалительной фазой, связанной с эндотелиальной активацией, и более поздней фиброзной фазой, проявляющейся коллагенизацией ткани и заметной толщиной кожи, их клинические проявления различаются.СЛ, как правило, ограничивается кожей и подкожным слоем, и хотя он не является смертельным, как ССД, до четверти пациентов могут иметь внекожные проявления заболевания, такие как артрит и увеит. Хотя при СС может поражаться любой орган, у детей чаще всего наблюдаются поражения сосудов (феномен Рейно), кожи (утолщение кожи), желудочно-кишечного тракта, легких и опорно-двигательного аппарата. Профили аутоантител при ССД в детском возрасте могут помочь в прогнозировании поражения внутренних органов. Лечение обеих форм склеродермии нацелено на активную воспалительную стадию и останавливает прогрессирование заболевания; тем не менее, все еще необходимо добиться прогресса в разработке более эффективной антифиброзной терапии, чтобы помочь обратить вспять повреждение, вызванное болезнью.
Ключевые слова: системный склероз, локализованная склеродермия, морфеа, детская ревматология
Детская склеродермия
Термин склеродермия буквально означает «склероз», склерозирование или уплотнение «дермы», кожи. «Склеродермия» охватывает обе формы заболевания: системный склероз (СС), характеризующийся фиброзом кожи, сосудов и внутренних органов, который чаще поражает взрослых, и локализованную склеродермию (ЛС), характеризующуюся фиброзом кожи и подлежащих тканей без сосудистых или поражение внутренних органов, которое чаще поражает детей.Хотя они имеют общую основную патофизиологию чрезмерного отложения коллагена при аутоиммунных заболеваниях, эти два состояния клинически различны с уникальными заболеваемостью и прогнозами (2). И то и другое редко встречается у детей: расчетная годовая заболеваемость СЛ составляет 1–3 случая на 100 000 детей [1], а ССД — 1 случай на миллион детей [2]. Средний возраст начала обеих форм детской склеродермии составляет от 7,3 до 8,8 лет. Хотя менее чем у 5% всех пациентов с ССД заболевание начинается в детском возрасте, у большинства пациентов с СЛ оно начинается в детском возрасте [3–6].К сожалению, существует значительная задержка в диагностике, в среднем от 1,9 до 2,8 лет для ССД [3, 4] и от 1,2 до 1,6 лет для СЛ [5, 7]. Для немногих случаев врожденного поражения СЛ среднее время установления диагноза больше, от 3,3 до 3,9 лет [7, 8]. Примерное соотношение женщин и мужчин при ССД у детей составляет 4:1 [3, 4], а при СЛ у детей — 2:1 [5]. Нет четких доказательств расовой предрасположенности к любой из форм детской склеродермии.
Хотя менее чем у 5% всех пациентов с ССД заболевание начинается в детском возрасте, у большинства пациентов с СЛ оно начинается в детском возрасте [3–6].К сожалению, существует значительная задержка в диагностике, в среднем от 1,9 до 2,8 лет для ССД [3, 4] и от 1,2 до 1,6 лет для СЛ [5, 7]. Для немногих случаев врожденного поражения СЛ среднее время установления диагноза больше, от 3,3 до 3,9 лет [7, 8]. Примерное соотношение женщин и мужчин при ССД у детей составляет 4:1 [3, 4], а при СЛ у детей — 2:1 [5]. Нет четких доказательств расовой предрасположенности к любой из форм детской склеродермии.
Детская склеродермия подразделяется на системную и локализованную болезнь, которая далее дифференцируется на подтипы на основании клинических проявлений поражения кожи.Клинические подмножества связаны с конкретными заболеваниями, как уже упоминалось.
ИЗЛ = интерстициальное заболевание легких; ЛАГ = легочная артериальная гипертензия; ROM = диапазон движений
Системный склероз
Классификация – клиническая и серологическая
Системный склероз (СС) является редким, но потенциально опасным для жизни состоянием. Существует три основных клинических подтипа, связанных с различными заболеваниями (): диффузный кожный (dc) SSc, характеризующийся широко распространенным и быстро прогрессирующим утолщением кожи (распространяющимся проксимальнее локтей и коленей), связанный с ранним поражением внутренних органов (легких, сердца и почек), ограниченный кожный ССД (lcSSc), характеризующийся ограниченным и непрогрессирующим утолщением кожи (ограничивается дистальными отделами конечностей), связанный с поздним заболеванием внутренних органов (легочная артериальная гипертензия, мальабсорбция), и перекрывающийся SSc, который может быть dcSSc или lcSSc с признаками другого заболевания соединительной ткани , такие как дерматомиозит или системная красная волчанка (СКВ) [9].Пациентов с CREST-синдромом (кальциноз, феномен Рейно, нарушение моторики пищевода, склеродактилия и телеангиэктазии) относят к ЛСКС. Некоторые пациенты с недифференцированным или смешанным заболеванием соединительной ткани могут иметь блестящую или полную кожу дистальных отделов пальцев, но у них должен быть основной критерий «уплотнение кожи или склероз проксимальнее пястной кости (MCP) или плюснефаланговой кости (MTP)».
Существует три основных клинических подтипа, связанных с различными заболеваниями (): диффузный кожный (dc) SSc, характеризующийся широко распространенным и быстро прогрессирующим утолщением кожи (распространяющимся проксимальнее локтей и коленей), связанный с ранним поражением внутренних органов (легких, сердца и почек), ограниченный кожный ССД (lcSSc), характеризующийся ограниченным и непрогрессирующим утолщением кожи (ограничивается дистальными отделами конечностей), связанный с поздним заболеванием внутренних органов (легочная артериальная гипертензия, мальабсорбция), и перекрывающийся SSc, который может быть dcSSc или lcSSc с признаками другого заболевания соединительной ткани , такие как дерматомиозит или системная красная волчанка (СКВ) [9].Пациентов с CREST-синдромом (кальциноз, феномен Рейно, нарушение моторики пищевода, склеродактилия и телеангиэктазии) относят к ЛСКС. Некоторые пациенты с недифференцированным или смешанным заболеванием соединительной ткани могут иметь блестящую или полную кожу дистальных отделов пальцев, но у них должен быть основной критерий «уплотнение кожи или склероз проксимальнее пястной кости (MCP) или плюснефаланговой кости (MTP)». считается несовершеннолетним SSc (). Существуют дополнительные два малых критерия, необходимые для диагностики ССД у детей в соответствии с международным консенсусным соглашением () [10].В дополнение к степени утолщения кожи определенные профили аутоантител в сыворотке, связанные со склеродермией, помогают идентифицировать подмножества ССД и прогнозировать их поражение органов как у взрослых, так и у детей с началом ССД [3, 4, 6, 11, 12]. Например, антитело против топоизомеразы (Scl-70) можно ожидать у пациента с dcSSc, и это может вызывать беспокойство в связи с быстрым прогрессированием поражения кожи и развитием интерстициального заболевания легких (ИЗЛ). [3, 4, 6]. Четырьмя наиболее распространенными антителами при ССД у детей являются антитела к топоизомеразе (Scl-70) (20–34%), обычно связанные с dcSSc и ИЗЛ, как уже упоминалось, за которыми следуют антитела к центромерам (ACA) (2–16%). связанный с lcSSc и легочной артериальной гипертензией (ЛАГ), а также антитела к U1-RNP и Pm-Scl (полимиозит-склеродермия) (2–16%), оба из которых обычно обнаруживаются у пациентов с перекрывающимися синдромами SSc и другими соединительными заболевания тканей с более выраженными признаками артрита и миозита, такие как дерматомиозит или системная красная волчанка [3, 4, 6].
считается несовершеннолетним SSc (). Существуют дополнительные два малых критерия, необходимые для диагностики ССД у детей в соответствии с международным консенсусным соглашением () [10].В дополнение к степени утолщения кожи определенные профили аутоантител в сыворотке, связанные со склеродермией, помогают идентифицировать подмножества ССД и прогнозировать их поражение органов как у взрослых, так и у детей с началом ССД [3, 4, 6, 11, 12]. Например, антитело против топоизомеразы (Scl-70) можно ожидать у пациента с dcSSc, и это может вызывать беспокойство в связи с быстрым прогрессированием поражения кожи и развитием интерстициального заболевания легких (ИЗЛ). [3, 4, 6]. Четырьмя наиболее распространенными антителами при ССД у детей являются антитела к топоизомеразе (Scl-70) (20–34%), обычно связанные с dcSSc и ИЗЛ, как уже упоминалось, за которыми следуют антитела к центромерам (ACA) (2–16%). связанный с lcSSc и легочной артериальной гипертензией (ЛАГ), а также антитела к U1-RNP и Pm-Scl (полимиозит-склеродермия) (2–16%), оба из которых обычно обнаруживаются у пациентов с перекрывающимися синдромами SSc и другими соединительными заболевания тканей с более выраженными признаками артрита и миозита, такие как дерматомиозит или системная красная волчанка [3, 4, 6]. Положительная доля пациентов с U1-RNP и Pm-Scl, вероятно, выше, чем сообщалось, поскольку некоторые авторы не включают случаи перекрывающегося SSc или смешанных заболеваний соединительной ткани в своих педиатрических когортах SSc. В целом, частота этих антител у детей отражает частоту клинической подгруппы dcSSc, lcSSc и перекрывающегося SSc [3, 4, 6]. По сравнению со взрослым SSc перекрывающийся SSc гораздо чаще встречается у детей (29% против 9%). , что отражается повышенной частотой антител U1-RNP и Pm-Scl и связано с более высоким процентом миозита и артрита, наблюдаемых у детей с ССД [3].Есть несколько других аутоантител, связанных с ССД, которые связаны с различными органными проявлениями; однако они гораздо реже встречаются при СД в детском возрасте. Одним из них является антитело к РНК-полимеразе III (POL3), которое относится к тяжелым почечным заболеваниям в форме склеродермического почечного криза. POL3 редко наблюдается при ССД в детском возрасте, но может быть обнаружен у 30% взрослых пациентов с ССД и отражает клинически значимую более высокую долю почечной недостаточности при ССД у взрослых (3, 6). Несмотря на то, что многие аутоантитела, связанные со склеродермией, были идентифицированы у взрослых ССД, существует высокая доля детей, 20-23%, которые имеют положительный результат АНА без идентифицированных специфических аутоантител [3, 6].
Положительная доля пациентов с U1-RNP и Pm-Scl, вероятно, выше, чем сообщалось, поскольку некоторые авторы не включают случаи перекрывающегося SSc или смешанных заболеваний соединительной ткани в своих педиатрических когортах SSc. В целом, частота этих антител у детей отражает частоту клинической подгруппы dcSSc, lcSSc и перекрывающегося SSc [3, 4, 6]. По сравнению со взрослым SSc перекрывающийся SSc гораздо чаще встречается у детей (29% против 9%). , что отражается повышенной частотой антител U1-RNP и Pm-Scl и связано с более высоким процентом миозита и артрита, наблюдаемых у детей с ССД [3].Есть несколько других аутоантител, связанных с ССД, которые связаны с различными органными проявлениями; однако они гораздо реже встречаются при СД в детском возрасте. Одним из них является антитело к РНК-полимеразе III (POL3), которое относится к тяжелым почечным заболеваниям в форме склеродермического почечного криза. POL3 редко наблюдается при ССД в детском возрасте, но может быть обнаружен у 30% взрослых пациентов с ССД и отражает клинически значимую более высокую долю почечной недостаточности при ССД у взрослых (3, 6). Несмотря на то, что многие аутоантитела, связанные со склеродермией, были идентифицированы у взрослых ССД, существует высокая доля детей, 20-23%, которые имеют положительный результат АНА без идентифицированных специфических аутоантител [3, 6].
Типичная блестящая и толстая кожа рук с уплотнением кожи, распространяющимся вверх по предплечью (за пястно-фаланговые суставы) у пациента с системной склеродермией.
Таблица 1
Временные рекомендации по классификации Критерии несовершеннолетних системного склероза *
| Основным критерием (обязательно) | Устранение кожи / утолщение проксимальных к MCP или MTP-соединениям | ||
| Незначительный критерий (2 требуется) (Scleroderma конкретный орган участие органов) | |||
| COSCORE | |||
| Sclerodactyly | |||
| Мопиллярные изменения ногтей (мегакапилларии и авашелярные зоны) | |||
| Digital Tip lexers | |||
| Dysphasia | |||
| Gastroesophageal Reffull | |||
| Cardiac | |||
| Arrhhythmias | |||
| Сердце сбой | |||
| почек | |||
| почек | |||
| Scleroderma Renal Crisis | |||
| новая артериальная гипертония | |||
| легочной артериальной гипертензии (первичной или вторичной по отношению к ILD, оценивали по данным эхокардиографии) | |||
| Неврологические | |||
| невропатии | |||
| кистевой туннельный синдром | |||
| Musculoskeletal | |||
| фрикционные трется Сухожильные | |||
| Arthrititis | |||
| серологический | |||
| Антиатомное антитело | |||
| SSC-селективный автоантибоди es | |||
| Антитопоизомераза 1 (Scl-70), антицентромерная, антиРНК-полимераза I или III, анти-PM-Scl, антифибриллин |
Клинические проявления
Основные клинические проявления у детей SSc
Сосудистый
Общими признаками в начале детского SSc являются феномен Рейно (RP) (70%) и кожные изменения рук (60%), включая отек, склеродактилию и уплотнение проксимальнее MCP [4].В наибольшей изученной когорте педиатрических ССД (153 пациента) 53% имели как уплотнение кожи рук, так и РН, а 10% пациентов с РН также имели инфаркты пальцев [4]. На протяжении всего заболевания почти у всех будет РН (97%) и изменения кожи кистей (96%) [3].
Девочка-подросток с типичной (А) склеродактилией и (В) пальцевыми изъязвлениями и язвами.
Феномен Рейно — вазоспастическая реакция, приводящая к трехфазному изменению цвета рук (сначала бледность из-за вазоконстрикции, затем посинение вследствие цианоза и, наконец, краснота из-за реперфузии с последующим отеком и болью), связанная с ощущением онемения и покалывания .RP также может возникать в пальцах ног и других акральных областях, включая уши, кончик носа или язык. При ССД артериолы более узкие и жесткие из-за выраженной гиперплазии интимы и фиброза вследствие эндотелиальной дисфункции, что приводит к более выраженному РП с последующим повреждением/ишемией тканей [13]. Эти васкулопатические изменения идентифицируются в капиллярном ложе ногтевого ложа у этих пациентов с помощью капиллярной микроскопии, демонстрируя дилатацию, извилистость, кровоизлияние, выпадение (аваскуляризацию) и более позднюю разветвленность капилляров [14].Сообщается, что распространенность капиллярных изменений ногтевого ложа при ССД у детей составляет 50% [3, 4], но, вероятно, выше, если проводилась стандартизированная микроскопия. Плохая перфузия пальцев в конечном итоге приводит к изъязвлениям кончиков пальцев, изъязвлению (2), а в более тяжелых случаях — к аутоампутации. Здоровые люди также могут страдать феноменом Рейно без риска развития основного заболевания соединительной ткани (первичный синдром Рейно). У этих людей обычно никогда не развиваются язвы на кончиках пальцев или капиллярные изменения ногтевого валика, поскольку у них нет основной васкулопатии.Признаки, которые помогают клиницисту дифференцировать первичный феномен Рейно от феномена, вторичного по отношению к заболеванию соединительной ткани (ЗСТ), как у детей, так и у взрослых, следующие: отсутствие изменений капилляров ногтевого валика, изъязвление и/или изъязвление кончиков пальцев и отрицательный АНА [15, 16].
;;9 и 2,8 года между первыми признаками заболевания и диагнозом [3, 4]. В начале клинического течения кожа отечна, особенно на дистальных отделах конечностей; редко присутствует поражение проксимальных отделов конечностей, лица и туловища. Фаза индурации, в честь которой названа склеродермия, характеризуется потерей естественной податливости кожи и наличием пальпируемой толщины кожи. Кожа приобретает блестящий, напряженный вид с сужением дистальных отделов пальцев (и ). Со временем, по мере того как кожа утолщается, а нижележащие структуры, такие как сухожилия, поражаются и укорачиваются, суставы пальцев начинают терять диапазон движений как при разгибании, так и при сгибании, а в более тяжелых случаях возникает деформация «когтеобразной руки», что сильно влияет на работоспособность. повседневная деятельность (ADL).Типичное лицо склеродермии с натянутой кожей и атрофией кожи характеризуется сморщенным носом, тонкими сжатыми губами, маленьким ртом, выступающими зубами и невыразительным видом (4). Толщина кожи оценивается как легкая, умеренная и тяжелая по всему телу, и кумулятивный балл получается с использованием модифицированной шкалы кожи Роднана (mRSS) [17]. mRSS получают в ходе лонгитюдных посещений, и можно рассчитать скорость прогрессирования толщины кожи (STPR), что помогает прогнозировать время поражения внутренних органов у пациентов с dcSSc.Высокий STPR связан со склеродермическим почечным кризом [18].
Невыраженное лицо при склеродермии. Девочка препубертатного возраста с уменьшенным ротовым отверстием и «поджатыми губами».
Другие кожные проявления при СС включают подкожные отложения кальция (кальциноз кожи), которые могут возникать в точках давления, обычно обнаруживаемых на разгибательных поверхностях суставов кистей при СС, и могут иногда выдавливаться через кожу подобно дерматомиозиту. Эти поражения могут быть болезненными и изъязвляться.Однако они чаще связаны с хроническим заболеванием и редко присутствуют во время начала заболевания и постановки диагноза. Телеангиэктазии также являются проявлением ССД, чаще всего встречаются в варианте lcSSc (ранее известном как CREST-синдром) и обычно локализуются на лице и верхних конечностях.
Другие органные проявления при ССД (в порядке убывания частоты) включают желудочно-кишечные, легочные, скелетно-мышечные, сердечные, почечные и неврологические симптомы. Желудочно-кишечные симптомы встречаются примерно у половины детей, хотя более детальное исследование часто указывает на наличие аномалий у большего процента [3].Нарушение моторики пищевода и гастроэзофагеальный рефлюкс часто приводят к дисфагии и эзофагиту. Дистальная дисфагия, особенно при приеме твердой пищи, вызывающая ощущение «застревания пищи» в середине грудной клетки, является типичной и представляет собой дисфункцию дистальной гладкой мускулатуры пищевода. Гастропарез с замедленным опорожнением желудка в дополнение к дисфункции пищевода может привести к усилению симптомов рефлюкса. Обычно пациенты жалуются на ночной кашель в положении лежа на животе из-за тихой аспирации и солоноватый привкус во рту при пробуждении из-за тихого рефлюкса.Хронический рефлюкс может привести к стриктурам пищевода. Оценка желудочно-кишечного тракта с использованием манометрии и внутрипищеводного 24-часового рН-мониторинга считается чувствительным индикатором сниженного тонуса пищевода и рефлюкса [19]. Однако в педиатрии эти методы используются реже. Вместо этого обычно используется исследование глотания для оценки аспирации и нарушений моторики с последующим исследованием верхних отделов желудочно-кишечного тракта с последующим исследованием тонкой кишки. Если поражена тонкая кишка, спазмы, диарея и запор могут быть результатом перистальтической дисфункции.Эпизоды псевдообструкции с постпрандиальным вздутием живота, болью и тошнотой могут возникать из-за функциональной кишечной непроходимости. Могут наблюдаться избыточный бактериальный рост, стеаторея, потеря веса, заворот и даже перфорация. Заболевание толстой кишки проявляется в виде широкоротых дивертикулов и потери нормальной архитектуры толстой кишки. Другим осложнением поражения ЖКТ при ССД является острое желудочно-кишечное кровотечение, связанное с эктазией антральных вен желудка (ГАВЭ), требующее фотокоагуляции. Это состояние редко встречается у детей и связано с ранним dc SSc, РНК-полимераза III положительных пациентов [20].
Поражение легких при педиатрическом ССД составляет в литературе от 30 до 70% и включает интерстициальное заболевание легких (ИЗЛ), легочную артериальную гипертензию (ЛАГ) (либо первичную, которая возникает в результате васкулопатии, либо вторичную из-за ИЗЛ), и аномальные легочные функциональные тесты (PFTs). Особое беспокойство вызывают аномалии PFT, такие как снижение форсированной жизненной емкости легких (ФЖЕЛ) и диффузионной способности монооксида углерода (DLCO) [21]. Интерстициальное заболевание легких (ИЗЛ) обычно начинается с воспалительной фазы, авеолита, характеризующегося смешанным клеточным инфильтратом в интерстиции легкого, который распространяется в альвеолярные пространства.Бронхоавеолярный лаваж (БАЛ) демонстрирует увеличение нейтрофилов и/или эозинофилов в дополнение к альвеолярным макрофагам [22]. На наличие авеолита также указывают затемнения по типу матового стекла на компьютерной томографии высокого разрешения (КТВР) легких. По мере прогрессирования ИЗЛ воспаление стихает, так как происходит переход в сторону фиброза с утолщением альвеолярных стенок и ремоделированием легкого. Прогрессирующий легочный фиброз может привести к рестриктивному заболеванию легких от умеренной до тяжелой степени со значительным снижением ФЖЕЛ и DLCO.Клинически ИЗЛ обычно проявляется в виде медленно прогрессирующей одышки при физической нагрузке в течение многих лет. Для сравнения, ЛАГ проявляется быстрым прогрессированием одышки при физической нагрузке в течение нескольких месяцев. В условиях ЛАГ PFT демонстрируют снижение DLCO и аномальные результаты эхокардиограммы с предполагаемым систолическим давлением в легочной артерии (ЛА) или правом желудочке (ПЖ)> 40 мм рт.ст., что подтверждается катетеризацией правых отделов сердца со средним давлением ЛА> 25 мм рт.ст. Для большинства детей с ССД или подозрением на ССД первоначальная оценка включает рентгенографию грудной клетки, тесты функции легких, включая DLCO, и эхокардиограмму.Дополнительные сердечно-легочные тесты проводятся, если обнаруживаются аномалии или пациент слишком молод, чтобы надлежащим образом завершить тестирование (например, PFT). Аномальные PFT требуют оценки с помощью HRCT грудной клетки и, возможно, BAL, если требуются дополнительные доказательства воспаления (авеолит) или необходима оценка инфекции перед началом терапии [21, 23]. Признаки ЛАГ на эхокардиограмме более формально оцениваются при катетеризации правых отделов сердца.
По сравнению с началом ССД у взрослых поражение опорно-двигательного аппарата чаще встречается у детей с ССД.Приблизительно 30–40% страдают воспалительным артритом (суставные выпоты) в дополнение к типичному «сухому синовиту» склеродермии, выражающемуся в фиброзе сухожилий, пересекающих суставы, что ограничивает диапазон их движений (ROM). В начале патологического процесса, особенно у пациентов с dcSSc, теносиновит/бурсит вызывает ощутимый шум трения сухожилий («кожистый хруст») при разгибании или сгибании сустава [24]. При миопатии у больного обычно отмечается симметричная проксимальная слабость, особенно плечевого пояса и плечевых мышц, иногда с выраженной атрофией.Миозит также проявляется повышением уровня мышечных ферментов и изменениями групп мышц на МРТ, сходными с таковыми при ювенильном дерматомиозите (ЮДМ). Биопсия мышц при ССД отличается от таковой при ЮДМ наличием большего количества фиброза и наличием утолщенных капилляров [25]. Дети с dcSSc и миозитом особенно подвержены риску тяжелого поражения сердца, включая дефицит перфузии миокарда и дилатационную кардиомиопатию [26].
Поражение сердца , хотя и редко, но является основной причиной связанной со склеродермией смертности у детей с ССД, о чем свидетельствуют 10 из 15 смертей в когорте Martini et al. [4] и 5 из 32 смертей в Scalapino et al. когорта [3].Сердечные проявления являются результатом сочетания миокардиального фиброза, сосудистой недостаточности и воспаления и включают сердечную блокаду, аритмию, застойную сердечную недостаточность, кардиомиопатию и перикардит. В отличие от детского ССД, смертность от склеродермии у взрослых в первую очередь обусловлена поражением легких, как ИЗЛ, так и ЛАГ [27].
Легкая почечная дисфункция не редкость при ССД и является вторичной по отношению к васкулопатии, а не к гломерулонефриту, который наблюдается при системной красной волчанке.Это подтверждается патологическими данными пролиферации интимы междольковых артерий почек и отсутствием активного мочевого осадка (т.е. эритроцитарных цилиндров). Напротив, более тяжелая и болезненная форма заболевания почек, склеродермия , почечный криз (SRC), встречается только примерно у 15% взрослых пациентов с ССД и еще реже у детей. СРК характеризуется ускоренной артериальной гипертензией, почечной недостаточностью, микроангиопатической гемолитической анемией и тромбоцитопенией.При SSc у взрослых SRC ранее имел 20% выживаемость в течение 1 года, но теперь выживаемость составляет 80% из-за появления ингибиторов ангиотензинпревращающего фермента (ACEI) [27]. SRC чаще всего наблюдается на ранних стадиях заболевания у пациентов с РНК-полимеразой III dcSSc и, как полагают, усиливается при более высоких дозах кортикостероидов. Почечный криз у детей с ССД встречается редко, с менее чем 5% случаев, и только 5 случаев зарегистрированы (2 из которых умерли от почечной недостаточности) среди трех больших опубликованных когорт [3, 4, 28].
Исход
Несмотря на все возможные поражения органов при ССД у детей, она имеет гораздо более благоприятный прогноз через 5, 10 и 15 лет по сравнению со ССД у взрослых с более низкой частотой тяжелого поражения органов. Foeldevari и коллеги [28] недавно сообщили, что показатели выживаемости Kaplan Meier в течение 10 лет для детей составляют 98% по сравнению с 75% у взрослых. Лечение ССД зависит от поражения органов, но общие меры, такие как антациды для защиты ЖКТ, прокинетики для нарушения моторики, чередование антибиотиков для мальабсорбции, а также сосудорасширяющие средства при феномене Рейно (RP) (могут также помочь при ЛАГ), нестероидные Обычно рекомендуются противовоспалительные препараты (НПВП) и физиотерапия (ЛТ) при артрите или тендините.Кортикостероиды при миозите, артрите и других воспалительных клинических проявлениях следует использовать с осторожностью, поскольку они могут спровоцировать склеродермический почечный криз. Более конкретные рекомендации по лечению различных органных проявлений были согласованы на консенсусной конференции международных экспертов по склеродермии и опубликованы в 2009 г. [29]. Примечательно, что в лечении раннего диффузного кожного заболевания как у детей, так и у взрослых произошел переход от использования D-пеницилламина к микофенолата мофетилу (ММФ) из-за потенциальных антипролиферативных эффектов, хотя это не было включено в рекомендации по лечению, которые обсуждались ранее. 30].
Локализованная склеродермия
Классификация
Локализованная склеродермия, также известная как «морфея», имеет другой тип поражения кожи, чем системный склероз (СС), и включает несколько подтипов, классифицированных по глубине и характеру поражения(й). Группа заболеваний характеризуется фиброзом, который в основном ограничивается кожей и подкожной клетчаткой; однако более глубокие формы также включают фасции, мышцы, сухожилия и суставную капсулу. Традиционная классификация, предложенная Peterson et al.[31] описали пять подклассов, включая бляшечную морфею, генерализованную морфею, буллезную морфею, линейную морфею, глубокую и пансклеротическую морфею (1). Однако совсем недавно консенсус экспертов внес изменения, чтобы предоставить более клинически применимые классификации, названные «Критерии Падуи», которые включают подтип «смешанного морфеа», учитывая, что распространенность этой категории составляет 15% () [32].
Таблица 2
| A: LS ° Подтипы: Mayo Классификация ‡ |
|---|
| Налет Morphea † |
| Morphea ан бляшек † |
| каплевидный Morphea |
| атрофодермия из Pasini и Пьерини |
| Обобщенная Morphea † |
| Буллезная Morphea |
| линейный Morphea † |
| линейная Morphea (линейная склеродермия) † |
| En купе де сабля † |
| Progressive гемифациальный атрофия |
| Deep Morphea † |
| Подкожный Morphea † |
| эозинофильный фасциит | Morphea profunda † |
| Отключения pansclerotic Morphea детей |
| б: Предлагаемые подтипов для ювенильного LS °: Падуя Предварительной Классификации ‡ | |||
|---|---|---|---|
| Описанного Morphea | Овальные/круглые поражения
| 1||
| Linear Scleroderma | Линейные поражения могут включать дерма, подкожная клетчатка, мышцы, кости
| Обобщенные Morphea | ≥ 4 Большие бляшки (> 3см) по крайней мере 2 7 анатомических областей: голова/шея, правая верхняя конечность, LUE, RLE, LLE, передняя часть туловища, задняя часть туловища | |
| Пансклеротическая морфеа | Окружное поражение конечности (за исключением кончиков пальцев рук и ног) Все глубины кожи/подкожной клетчатки /мышца/кость | ||
| Смешанная форма морфеа | Комбинация двух или более вышеперечисленных подтипов | ||
Активность заболевания и признаки повреждения (клинические и гистологические данные, клинические и гистологические признаки) Сходны с СЛ
более ранняя более активная фаза заболевания и более поздняя более фиброзная фаза ().Ранние активные поражения при СЛ характеризуются фиолетовой воспалительной каймой или «сиреневым кольцом» (1) и индурацией кожи по всему очагу поражения, включая границу. Во время этого активного состояния поражения также могут увеличиваться, и могут накапливаться новые поражения [33]. Гистологическая картина на ранних стадиях заболевания состоит из периваскулярного инфильтрата преимущественно из лимфоцитов с примесью редких плазматических клеток и эозинофилов в глубоком ретикулярном слое дермы и подкожной клетчатке, сопровождающемся утолщением коллагеновых пучков, уменьшением эластических волокон и набуханием эндотелиальных клеток [34, 35].С течением времени поражение болезнью накапливается и проявляется увеличением толщины кожи, особенно в центре поражения, иногда оставляя склеротический очаг цвета слоновой кости (). Гистологическая оценка кожи выявляет утолщенный гипоцеллюлярный (гомогенизированный) коллаген, который заменил прежний воспалительный инфильтрат вокруг придатков дермы, оставив после себя атрофические эккринные железы и волосяные фолликулы. Кроме того, уменьшается количество капилляров с фиброзной стенкой и суженным просветом [34, 35].Клинический результат этих находок отражается в кожной и подкожной атрофии. При физикальном осмотре, когда дерма атрофична, наблюдается отсутствие роста волос в месте поражения, видимые вены и легкий вид «обрыва» на коже (). Подкожная атрофия существует в непрерывном диапазоне и может быть как легкой, как уплощение кожи, так и более «выемчатым» вогнутым видом жировой ткани при более умеренных поражениях, а в самых тяжелых случаях до такой степени, что можно увидеть мышцы, движущиеся под кожей.Поствоспалительная гипер- и гипопигментация также являются результатом предшествующего воспаления и часто являются признаками, которые привлекают внимание врача к поражению. Часто семьи пропускают раннюю активную фазу и считают эритему/фиолетовый цвет скорее синяком или травмой, но именно тот факт, что «синяк, который не проходит» (поствоспалительная гиперпигментация), часто побуждает семью искать медицинская помощь ().
Фазы локализованной склеродермии и связанные с ней признаки.Признаки активного заболевания, эритема, уплотнение/отек кожи и новые или увеличивающиеся очаги поражения возникают первыми и уступают место признакам поражения заболеванием, диспигментации, атрофии и утолщению кожи.
Типичное «сиреневое кольцо», наблюдаемое на границе множественных бляшек на туловище, указывающее на активное заболевание, любезно предоставлено Еленой Поуп из Children’s Sick Kids, Торонто.
Восковидно-желтый очаг поражения в центре бляшки, остающийся на правой стороне шеи спустя годы после начала локализованной склеродермии.
Кожная атрофия LS, проявляющаяся в виде «капельных» поражений у девочки-подростка с генерализованной морфеей.
Патогенез (общий для ССД и ССД)
Общие гистологические данные на коже пациентов с ССД и СЛ иллюстрируют их общую патофизиологию, несмотря на различные клинические особенности заболевания. Оба являются аутоиммунными заболеваниями с воспалительным, фиброзным и сосудистым компонентом. Популяции мононуклеарных клеток, идентифицированные как в образцах кожи SSc, так и в LS, представляют собой преимущественно Т-клетки, как CD8, так и CD4-положительные, с более высоким процентом клеток CD4 (T-хелперы) [36, 37]. Ассоциированные с Th-клетками цитокины и хемокины, такие как интерлейкины [IL]-2, -4, -6, -8, -13 и TNF-a, были идентифицированы в циркулирующей сыворотке и стимулированных PBMC у пациентов с LS и SSc [38–38]. 43].Следует отметить, что некоторые сывороточные цитокины были обнаружены в более высоких концентрациях у пациентов с LS по сравнению с СС, что интересно, поскольку LS называют «локализованным заболеванием»; однако это может подтверждать высокую частоту внекожных проявлений (примерно 25%), продемонстрированную при СЛ [44]. Было выявлено несколько корреляций между клиническими переменными тяжести заболевания LS и цитокинами и их растворимыми рецепторами, такими как количество бляшек и линейных поражений (IL-13, TNF-a, sIL-2r, sIL-6r), генерализованная морфия и субпопуляции линейной склеродермии (IL-2,-4,-6,-8,-13, TNF-a, sIL-2r, sIL-6r) и корреляция антител, антигистоновые и/или оц-ДНК антитела (IL -4, -6, TNF-a, sIL-2r, sIL-6r) [38–40, 45–47].Эти ассоциированные с Т-клетками цитокины, в свою очередь, дополнительно стимулируют фибробласты и эндотелиальные клетки к выработке трансформирующего фактора роста бета (TGF-β) и фактора роста соединительной ткани (CTGF), которые считаются основными стимуляторами тканевого фиброза (за счет увеличения коллагена). продукция) и повреждение эндотелиальных клеток (через влияние молекул адгезии ICAM-1 и VCAM-1) [48–51].
Микрохимеризм является еще одной интересной областью, изучаемой в отношении общего патогенеза LS и SSc.Клинически и гистологически LS и SSc имеют общие характеристики с хронической болезнью «трансплантат против хозяина» (cGVHD) (1), типом микрохимеризма, который играет роль в патогенезе. Химерные клетки были идентифицированы у пациентов с LS и SSc с использованием методов ПЦР в реальном времени [52, 53].
Линейное поражение туловища (A) и ног (B) у ребенка раннего возраста со склеродерматозной хронической болезнью «трансплантат против хозяина».
Аутоиммунитет, как один из компонентов распространения LS, подтверждается профилем аутоантител, сопутствующими сопутствующими аутоиммунными заболеваниями и семейным анамнезом аутоиммунных заболеваний, наблюдаемых у пациентов с LS.Аутоантитела обычно положительны при LS, включая антиядерные антитела (ANA), антигистоновые антитела (AHA) и антитела к одноцепочечной ДНК (ss-DNA Ab) [54]. ANA является классическим серологическим маркером аутоиммунного заболевания и обнаруживается у пациентов с СЛ с высокой частотой, в диапазоне от 42 до 73% [5, 55]. Считается, что более высокие титры связаны с ранним развитием заболевания [55] и повышенным риском внекожных проявлений [44, 56]. Распространенность оц-ДНК при СЛ составляет примерно 50% [57] и связана с линейной склеродермией (особенно с контрактурами суставов), вовлечением глубоких мышц и увеличением количества анатомических участков; все это отражает увеличение тяжести заболевания [56, 58].Было показано, что в подгруппе пациентов с LS одноцепочечное антитело к ДНК отражает статус активности заболевания, при этом титры снижаются по мере клинического улучшения кожных поражений [54, 56]. АГА встречается с аналогичной частотой при СЛ (47%) [59] и также коррелирует с показателями тяжести заболевания; включая общую площадь вовлеченной поверхности тела, количество поражений, линейное заболевание и наличие контрактур суставов [54, 56]. В исследовании, посвященном линейной склеродермии, сочетание положительных ss-ДНК и AHA было обнаружено у 23% пациентов (взрослых и детей с СЛ) и достоверно коррелировало с наличием контрактур суставов [56].Вышеупомянутые антитела указывают на тяжесть заболевания и могут помочь в стратификации пациентов с ранним диагностированным заболеванием, предполагая возможное начало более агрессивной терапии и усиленное наблюдение.
Сопутствующие и семейные ревматические и аутоиммунные заболевания более распространены при СЛ (детском и взрослом) по сравнению с общей популяцией [60]. Сопутствующее заболевание было зарегистрировано у 5–10% детей [7, 44, 60] и 30% взрослых с СЛ [60]. Наиболее распространенными заболеваниями были дерматологические аутоиммунные заболевания (псориаз, витилиго, очаговая алопеция), за которыми следовали ревматические состояния (ювенильный идиопатический артрит, ревматоидный артрит (РА), системная красная волчанка (СКВ) и синдром Шегрена) [7, 60].При оценке по подтипу СЛ частота сопутствующего аутоиммунного заболевания была значительно выше у пациентов с генерализованным морфеозом [60]. Семейные ревматические заболевания и другие аутоиммунные заболевания были зарегистрированы у 12-25% всех пациентов (как у детей, так и у взрослых), наиболее частым из которых был псориаз, за которым следовали РА, СКВ, ССД, СЛ, рассеянный склероз и тиреоидит [5, 7]. , 60]. Вновь была продемонстрирована связь между генерализованной морфеей и повышенной частотой этих заболеваний [5].
Клинические проявления
Хотя термин «локализованная склеродермия» или «морфея» объединяет все подтипы в одну категорию, их кожные/подкожные проявления и внекожные ассоциации совершенно уникальны. Линейная склеродермия (LiScl) является наиболее распространенным подтипом, встречающимся у 50–60% детей с LS, и характеризуется линейной полосой или полосой, поражающей дерму, подкожную клетчатку и часто подлежащие мышцы, сухожилия и кости. LiScl обычно возникает в виде одиночной линейной односторонней полосы на конечностях, туловище или голове (5, 7).Наиболее часто поражаются ноги, за ними следуют руки, лобная часть головы и туловище. Поражения LiScl, поражающие конечности, вызывают наибольшее беспокойство, когда они пересекают сустав, поскольку существует вероятность склероза и уплотнения нижележащих мышц, сухожилий и суставной капсулы, что приводит к контрактуре сустава и связанной с этим дисфункции конечностей. Особое беспокойство у детей с LiScl вызывает поражение эпифизарной зоны роста, что может привести к необратимому укорочению или атрофии конечности.Ортопедические осложнения регистрируются примерно у 30–50% пациентов с LiScl конечностей (конечностей) [7, 58, 61]. Peterson и соавторы сообщили о легкой или умеренной инвалидности при последующем наблюдении у пациентов с LiScl конечностей в их когорте (среднее время наблюдения 9,2 года) [1].
Линейная склеродермия, затрагивающая одностороннее поражение правой верхней конечности (А). У этой молодой женщины ассоциированные контрактуры лучезапястных, пястных и фаланговых суставов. Суставные контрактуры кисти отчетливо видны по сравнению с контралатеральной стороной при активном разгибании (В) и сгибании (С).
Приблизительно у 25% пациентов с LiScl наблюдается поражение волосистой части головы [5]. Линейные поражения кожи головы и лба называются «удар саблей» (ECDS), потому что линейная депрессия напоминает шрам от саблевидной раны. Прогрессирующая гемифациальная атрофия, также известная как синдром Парри-Ромберга (PRS), представляет собой еще один линейный вариант LS, вызывающий гемиатрофию лица, включая нижнюю, верхнюю челюсть, язык, подкожную и мышечную ткани (4). Как правило, ECDS ассоциируется с кожными проявлениями диспигментации и толщины кожи, тогда как PRS не является и обычно имеет более выраженную подкожную атрофию.Эти расстройства могут возникать вместе, либо в одном и том же месте (сторона лица), либо на противоположных сторонах [62]. Несмотря на противоречивость, ECDS и PRS, вероятно, представляют собой разные спектры одного и того же состояния, что подтверждается сходными проявлениями и преобладанием неврологической симптоматики [62, 63] и клеточных инфильтратов (лимфоцитарный инфильтрат сосудисто-нервных пучков) кожи [64] и ткани головного мозга [65]. ]. PRS и ECDS несут одинаковый риск неврологического поражения, при этом у 8–20% пациентов с этими состояниями наблюдается поражение центральной нервной системы, выражающееся в судорогах, хронических головных болях, неврите зрительного нерва и, реже, в нервно-психических изменениях или ишемическом инсульте [62, 63]. ].Также сообщалось об ортодонтических и офтальмологических аномалиях с одинаковой частотой при обоих состояниях [63]. Если начало заболевания (ECDS или PRS) происходит в возрасте до 10 лет, существует особый риск скелетной гипоплазии пораженной стороны лица из-за незрелости скелета, что приводит к стоматологическим и косметическим аномалиям [66]. ].
Саблезубое поражение (ECDS), поражающее волосистую часть черепа и голову односторонним образом (A), вызывающее умеренную потерю подкожной ткани и деформацию скелета (B).
Тяжелый пример синдрома Парри-Ромберга/гемифациальная атрофия правого лица.
Поверхностная ограниченная морфия, ранее называемая бляшечной морфеей по классификации Петерсона, является наиболее частым проявлением СЛ у взрослых (60%) и составляет примерно одну четверть СЛ у детей [5, 60]. Он характеризуется одной или несколькими овальными или округлыми участками уплотнения с восковым центром цвета слоновой кости и фиолетовым ореолом по периферии диаметром от 1 до 30 см. Эти поражения обычно возникают на туловище или проксимальных отделах конечностей (4).Поверхностный вариант ограничен эпидермисом и дермой. Глубокая ограниченная морфеа, ранее называемая подкожной морфеей, глубокой морфеей или глубокой морфеей, представляет собой круглое/овальное поражение, расположенное глубже в ткани. Эти поражения поражают дерму и подкожную клетчатку с различным проникновением в фасции и мышцы. Как правило, имеются только тонкие кожные изменения (такие как небольшая эритема и истончение дермы), но присутствует большое количество воспалительных инфильтратов в глубоких подкожных тканях и фасциях, вызывающих депрессию в жировой ткани («выемчатый» вид) и утолщение. , стянутая кожа.
Типичное изолированное овальное поражение на туловище, соответствующее поверхностным очерченным морфеям.
Генерализованная морфеа (ГМ) определяется наличием более 3 бляшек, каждое из которых имеет ширину более 3 см. Обычно бляшки сливаются и поражают несколько анатомических областей, чаще всего на туловище. ГМ — редкий подтип, встречающийся менее чем у 10% пациентов с СЛ [5, 7]. Как упоминалось ранее, этот подтип часто связан с несколькими аутоиммунными явлениями, включая антитела к одноцепочечной ДНК и AHA, а также сопутствующий и семейный анамнез аутоиммунных заболеваний.Системные симптомы, такие как миалгия, артралгия и утомляемость, чаще встречаются при ГМ [7, 60].
Пансклеротическая морфеа — редкая разновидность СЛ, поражающая только 1–2% пациентов и обычно приводящая к инвалидности. Первоначально он поражает конечности, но позже может мигрировать на туловище, не затрагивая только лицо и дистальные области пальцев рук и ног (4). Отмечается быстро прогрессирующий фиброз глубоких слоев дермы, подкожной клетчатки, фасций и мышц, иногда вовлекающий кости. Это приводит к значительной контрактуре суставов, атрофии мышц, изъязвлениям кожи и иногда к рестриктивной дыхательной недостаточности, если задействован грузовик.Развитие плоскоклеточного рака является дополнительным осложнением при пансклеротической морфее, особенно в области хронических ран [67].
Пансклеротическая морфеа, поражающая конечности по окружности и не затрагивающая дистальные отделы пальцев ног и пальцев.
Смешанная форма морфеа является последним подтипом, рассмотренным на основе консенсуса Падуи, и определяется наличием более чем одного клинического подтипа у одного пациента с СЛ. Смешанная форма морфеа довольно распространена и наблюдается примерно у 15% детей с СЛ.Наиболее частым проявлением смешанного морфеа является сочетание линейного и бляшечного морфеа [5].
Буллезный морфеоз иногда считают редким подтипом СЛ. Тем не менее, многие эксперты согласны с тем, что буллезные поражения являются вторичными по отношению к агрессивному линейному или глубокому СЛ и, вероятно, отражают нарушение лимфатического оттока. Эозинофильный фасциит (ЭФ), когда-то считавшийся типом глубокого морфеа по критериям Петерсона, не считается подтипом СЛ по критериям Падуи. Он часто ведет себя как смесь глубокой морфеи и линейной склеродермии, что приводит к глубокой инфильтрации и контрактурам суставов [31].)
Внекожные проявления
Внекожные проявления (ВКМ) нередки при СЛ и возникают примерно у четверти пациентов на протяжении всего заболевания [44]. Согласно большому международному исследованию 750 детей с LS, внеклеточное маточное окошко чаще встречается у пациентов с LiScl и в основном состоит из ортопедических осложнений (19% в целом в группе, 50% у детей с LiScl конечностей), неврологических или глазных нарушений (5% всей группе, 40% у лиц с LiScl головы), феномен Рейно (2.1% всей группы) и другие аутоиммунные состояния, такие как тиреоидит (2% всех подтипов) [44]. Менее распространенными ЭКМ являются желудочно-кишечные, респираторные и почечные, все они встречаются менее чем у 2% из 750 обследованных пациентов [44]. Эти оценки могут быть ниже, чем истинный процент ECM, поскольку в анализ были включены только пациенты с симптомами. Другим ограничением исследования было отсутствие контрольной популяции для оценки распространенности этих проявлений в сопоставимой детской популяции.В другом исследовании, в котором изучались желудочно-кишечные проявления у пациентов с СЛ (такие как гастроэпофагеальный рефлюкс) и изменения неврологической визуализации у пациентов с ECDS/PRS, исследователи обнаружили более высокую частоту внеклеточного матрикса; 20% и 60% соответственно [63, 68]. Интересно, что в исследовании Zulian et al., включавшем 750 детей с LS, из 168 пациентов с внеклеточным матриксом у 25% были суставные, неврологические и глазные проявления, не связанные с местом поражения кожи, т.е. артрит периферического сустава с непораженной вышележащей кожей и подкожной клетчаткой. , что еще раз подтверждает гипотезу о том, что «локализованная склеродермия» является более системным процессом [44].У 24 из 750 детей (3,2%) было выраженное поражение глаз, у большинства из которых была линейная склеродермия головы (67%) [69]. Эти результаты подтверждают рутинный офтальмологический скрининг пациентов с LS на наличие признаков воспаления глаз, особенно у пациентов с линейной склеродермией головы. Этот безопасный инструмент скрининга может выявить клинически «тихую» болезнь, а лечение может предотвратить долгосрочные последствия для зрения.
Оценка
Ни один лабораторный тест не является диагностическим для LS. Несмотря на «локализованное» заболевание, было выявлено несколько аутоантител, связанных с LS, которые могут помочь в диагностике и указать на более тяжелое/вовлеченное заболевание (, см. раздел об аутоиммунитете ).В частности, положительность ANA, AHA и одноцепочечной ДНК обнаруживалась с относительно высокой частотой (40–50%). Кроме того, ревматоидный фактор присутствует у трети больных, особенно у больных ГМ, а также коррелирует с тяжестью заболевания (количеством поражений) [5]. Общие маркеры воспаления, включая скорость оседания эритроцитов и уровень сывороточного иммуноглобулина, могут быть полезными маркерами активности заболевания у отдельных пациентов с СЛ, особенно с глубокими или эозинофильно-подобными вариантами.Однако у большинства пациентов с активными поражениями LS эти маркеры не повышены. Диагноз LS обычно ставится при клиническом обследовании, и, если есть сомнения, биопсия кожи и подкожной клетчатки может помочь в диагностике. Дифференциальная диагностика ограничена и включает два состояния кожи с хорошо выраженными бляшечными поражениями: склерозирующий и атрофический лихен и атрофодермию Пазини и Пьерини. Оба являются поверхностными дермальными и эпидермальными воспалительными состояниями, которые, по мнению некоторых, относятся к более мягкому спектру локализованной склеродермии.В дифференциальный диагноз включают кожную Т-клеточную лимфому, особенно с более глубоким багровым изменением цвета кожи, и коллагеному, обычно проявляющуюся уплотнением кожи и подкожного слоя без пигментных изменений с рождения или в раннем детстве. Другие возможные состояния связаны с определенным воздействием и, как правило, имеют более широко распространенные поражения, такие как реакция «трансплантат против хозяина» (состояние после трансплантации), нефрогенная фиброзирующая дерматопатия (гадолиний вводят при нарушении функции почек) и поздняя кожная порфирия (воздействие солнца).
Для оценки поражений кожи и подкожной клетчатки при LS используется несколько инструментов, таких как клиническая оценка кожи, инструменты измерения и визуализация.Клинические оценки кожи обычно используются врачами и включают модифицированную шкалу Роднана [17] и инструмент оценки локализованной склеродермии кожи (LoSCAT), который объединяет параметры активности заболевания и повреждения [33, 70]. Компьютеризированная оценка кожи (CSS) может использоваться для объективного определения размера поражения [71]. Инструменты измерения, в том числе дюрометр, который измеряет степень твердости кожи [72], и кутометр, который измеряет эластичность кожи [73], также были предложены в качестве полезных.Кроме того, для диагностики или оценки можно использовать несколько типов визуализации, таких как термография [74, 75], лазерная допплеровская флоуметрия [76], УЗИ [77, 78] и МРТ [79]. Некоторые из этих методов были проверены в LS, включая LoSCAT, CSS, ультразвук с использованием более высоких частот (10–25 МГц) и МРТ. Однако в настоящее время стандартные меры широко не используются, и оценка зависит от методов, специфичных для центра, которые зависят от опыта исследователя.
Недавние усилия Альянса по исследованиям детского артрита и ревматологии (CARRA) были сосредоточены на совместных проектах исследователей детской ревматологии и дерматологии в Северной Америке.Их цель состоит в том, чтобы установить конкретные клинические меры для оценки активности и повреждения, которые помогут в мониторинге прогрессирования и состояния болезни при LS. Эти проекты находятся в процессе оценки клинических показателей, таких как LoSCAT, и определения других с помощью многоцентровых исследований с группой клинических и ультразвуковых исследований локализованной склеродермии (LOCUS). Группа LOCUS состоит из экспертов по СЛ, как детских ревматологов, так и дерматологов, которым было предложено ранжировать клинические переменные, наиболее сильно отражающие активность заболевания и повреждения при СЛ.Консенсусное согласие >75% и высокая содержательная достоверность (модифицированная каппа-статистика 0,88–1,00) в отношении четырех параметров активности; появление новых поражений, расширение существующих поражений, эритематозный/фиолетовый оттенок на границе и уплотнение кожи на границе поражения [33]. Параметры повреждения кожных заболеваний, получившие наивысшее место среди группы, включали поствоспалительную гиперпигментацию и гипопигментацию, кожную и подкожную атрофию (согласие 88% и межпунктовый индекс достоверности 0.88 – 1,0) [70].
После начала системной иммуносупрессивной терапии у пациентов с умеренным и тяжелым СЛ признаки активности заболевания обычно исчезают в течение нескольких месяцев, а некоторые признаки повреждения уменьшаются от легкой до умеренной степени [80–85]. Именно после этого начального улучшения важно выявление субклинического заболевания, чтобы не отменять иммуносупрессию слишком рано, что может привести к рецидиву [80, 83, 84]. Недавнее исследование педиатрического СЛ выявило 28% рецидивов после средней продолжительности лечения метотрексатом, равной 2.4 года. Было обнаружено, что линейный подтип конечностей и пожилой возраст в начале заболевания являются значимыми предикторами рецидива, и, хотя это не значимо, была заметная разница в продолжительности лечения метотрексатом между рецидивирующими и нерецидивирующими пациентами [85]. Тем не менее, по-прежнему трудно обнаружить субклиническое заболевание, поэтому используются различные методы визуализации и тепловизионного исследования.
Лечение
Подход к лечению СЛ изначально зависит от тяжести заболевания, которое характеризуется подтипом СЛ, возможным вовлечением более глубоких тканей и структур (таких как суставы), локализацией СЛ и степенью активности заболевания.Продолжительность заболевания действительно влияет на состояние активности болезни, при этом большинство поверхностных ограниченных или бляшечных поражений «выгорают» через три-пять лет. Любой оставшийся центральный склероз может со временем смягчиться без вмешательства. Если остаются только параметры поражения болезнью и нет расширения, эритемы или других клинических параметров активности заболевания, допустимо простое наблюдение за поражением. Однако, если есть какие-либо признаки активного заболевания, как детские ревматологи, так и дерматологи согласны с тем, что одно поверхностное ограниченное поражение в не косметически значимом месте можно лечить либо местно кортикостероидами, ингибиторами кальциневрина, имиквимодом, витамином D, либо с помощью рецидивирующей фототерапии. с ультрафиолетовым (УФ) светом.Все методы имели клинический успех в серии случаев или в неконтролируемых испытаниях, что проявлялось уменьшением гиперпигментации и уменьшением толщины кожи. Многие дерматологи назначают эти препараты в комбинации и рекомендуют использовать окклюзионную повязку для увеличения всасывания местной терапии [86]. УФ-обработка имеет несколько модальностей; широкополосный UVA, узкополосный UVB и UVA-1. UVA-1 был успешным в двух рандомизированных контролируемых исследованиях, особенно в отношении ранних воспалительных и поверхностных кожных поражений [87, 88].Однако доступ к УФ-терапии ограничен, а лечение требует много времени (часто требуется три сеанса по 30–60 минут в неделю), что делает этот вариант сложным для многих. Кроме того, риск потенциальных долгосрочных эффектов, таких как старение кожи и канцерогенез, у детей неизвестен.
Среди ревматологов и дерматологов растет консенсус в отношении того, что системная терапия оправдана при умеренном или тяжелом СЛ. Признаки, которые могут указывать на среднетяжелое или тяжелое заболевание, включают глубокое вовлечение подкожной клетчатки, фасций и мышц, пересекающих сустав и потенциально вызывающих функциональные нарушения, линейные или атрофические поражения, поражающие лицо/кожу головы, быстро прогрессирующее или широко распространенное активное заболевание, а также неэффективна местная или УФ-терапия [86, 89].Наиболее часто используемой системной терапией является метотрексат (МТ) в сочетании с кортикостероидами (КС), что подтверждено недавним опросом 158 педиатрических ревматологов Северной Америки, проведенным Li et al. [89]. Клинические виньетки были розданы клиницистам, описывающим пациентов с умеренно-тяжелым LS, и клиницистов попросили дать свои рекомендации по лечению. Реакция клиницистов на начальную терапию заключалась в подавляющем большинстве случаев в комбинации с метотрексатом и кортикостероидами (более 80% использовали бы оба препарата). Несмотря на то, что было достигнуто отличное согласие в отношении выбора лекарств, схемы лечения широко различались в отношении приема (пероральный или комбинированный).подкожно или внутривенно), а также снижение дозы кортикостероидов и продолжительность терапии [89].
Подкомитет CARRA вырабатывает наилучшие согласованные протоколы лечения для пациентов с умеренным и тяжелым LS, чтобы помочь решить эту проблему и сузить список схем, которые были бы полезны клиницисту. Эти протоколы имеют четыре плана лечения для индукции терапии. Три протокола включают подкожное введение метотрексата (1 мг/кг, максимальная доза 25 мг в неделю), а четвертый был разработан для тех, у кого метотрексат неэффективен или у кого непереносимость метотрексата.Протоколы подразделяются на монотерапию метотрексатом, метотрексат с пероральным КС и постепенной дозировкой, метотрексат с внутривенным КС и микофенолата мофетил (ММФ) в дополнение к пероральному или внутривенному КС. Эти протоколы в настоящее время представляются для публикации. Было показано, что MMF эффективен при тяжелом резистентном к метотрексату педиатрическом LS, о чем свидетельствует серия случаев из 10 пациентов (два с пансклеротическим морфеозом, три с GM, два с LiScl конечностей и 3 с LiScl головы). Во всех случаях наблюдалось улучшение при приеме ММФ и удалось снизить дозу кортикостероидов [90].
Первое двойное слепое рандомизированное контролируемое исследование у детей с LS с использованием объективных показателей было проведено Zulian et al., в котором сравнивались метотрексат и пероральные кортикостероиды с метотрексатом и пероральными кортикостероидами.плацебо и пероральные КС. Было установлено, что метотрексат эффективен и безопасен, что подтверждает поддержку сообществом использования метотрексата при LS. Кроме того, у тех, кто лечился метотрексатом, значительно улучшилось состояние и было значительно меньше рецидивов по сравнению с теми, кто принимал только КС [84].
Другие методы лечения
Интенсивная физиотерапия и трудотерапия в сочетании с системной иммуносупрессивной терапией рекомендуется пациентам с линейной или глубокой склеродермией конечностей, чтобы помочь избежать или свести к минимуму контрактуры суставов.Кроме того, для пациентов с ECDS и / или PRS вмешательство пластической хирургии считается разумным после того, как заболевание находится в стадии ремиссии без каких-либо признаков клинической активности. Аугментация включает в себя растирание жировой ткани микрокаплями из подкожной клетчатки живота пациента, чтобы помочь заполнить часть подкожной атрофии, чтобы получить более симметричный контур лица (). При более тяжелом заболевании кожно-фасциальные лоскуты и реконструкция лицевых костей также являются вариантами, хотя обычно их применяют в позднем подростковом возрасте, после того как скелет созрел [91].Один центр сообщает, что из 10 подростков с ECDS и/или PRS, перенесших хирургическое вмешательство для коррекции лицевой деформации, 9 сообщили об удовлетворении результатами процедуры (процедур), и все они рекомендовали бы хирургическое вмешательство другим людям с этим заболеванием, что означает потенциальную большую пользу. влияние на качество жизни этих пациентов [92].
Синдром Парри-Ромберга, поражающий левое лицо до (А) и после (В) пересадки подкожно-жировой клетчатки.
Детская склеродермия – системная и локализованная формы
Краткое изложение
Детская склеродермия включает две основные группы клинических форм: системный склероз (СС) и локализованную склеродермию (ЛС).Хотя оба имеют общую патофизиологию с начальной воспалительной фазой, связанной с эндотелиальной активацией, и более поздней фиброзной фазой, проявляющейся коллагенизацией ткани и заметной толщиной кожи, их клинические проявления различаются. СЛ, как правило, ограничивается кожей и подкожным слоем, и хотя он не является смертельным, как ССД, до четверти пациентов могут иметь внекожные проявления заболевания, такие как артрит и увеит. Хотя при СС может поражаться любой орган, у детей чаще всего наблюдаются поражения сосудов (феномен Рейно), кожи (утолщение кожи), желудочно-кишечного тракта, легких и опорно-двигательного аппарата.Профили аутоантител при ССД в детском возрасте могут помочь в прогнозировании поражения внутренних органов. Лечение обеих форм склеродермии нацелено на активную воспалительную стадию и останавливает прогрессирование заболевания; тем не менее, все еще необходимо добиться прогресса в разработке более эффективной антифиброзной терапии, чтобы помочь обратить вспять повреждение, вызванное болезнью.
Ключевые слова: системный склероз, локализованная склеродермия, морфеа, детская ревматология
Детская склеродермия
Термин склеродермия буквально означает «склероз», склерозирование или уплотнение «дермы», кожи.«Склеродермия» охватывает обе формы заболевания: системный склероз (СС), характеризующийся фиброзом кожи, сосудов и внутренних органов, который чаще поражает взрослых, и локализованную склеродермию (ЛС), характеризующуюся фиброзом кожи и подлежащих тканей без сосудистых или поражение внутренних органов, которое чаще поражает детей. Хотя они имеют общую основную патофизиологию чрезмерного отложения коллагена при аутоиммунных заболеваниях, эти два состояния клинически различны с уникальными заболеваемостью и прогнозами (2).И то и другое редко встречается у детей: расчетная годовая заболеваемость СЛ составляет 1–3 случая на 100 000 детей [1], а ССД — 1 случай на миллион детей [2]. Средний возраст начала обеих форм детской склеродермии составляет от 7,3 до 8,8 лет. Хотя менее чем у 5% всех пациентов с ССД заболевание начинается в детском возрасте, у большинства пациентов с СЛ оно начинается в детском возрасте [3–6]. К сожалению, существует значительная задержка в диагностике, в среднем от 1,9 до 2,8 лет для ССД [3, 4] и от 1,2 до 1,6 лет для СЛ [5, 7].Для немногих случаев врожденного поражения СЛ среднее время установления диагноза больше, от 3,3 до 3,9 лет [7, 8]. Примерное соотношение женщин и мужчин при ССД у детей составляет 4:1 [3, 4], а при СЛ у детей — 2:1 [5]. Нет четких доказательств расовой предрасположенности к любой из форм детской склеродермии.
Детская склеродермия подразделяется на системную и локализованную болезнь, которая далее дифференцируется на подтипы на основании клинических проявлений поражения кожи. Клинические подмножества связаны с конкретными заболеваниями, как уже упоминалось.
ИЗЛ = интерстициальное заболевание легких; ЛАГ = легочная артериальная гипертензия; ROM = диапазон движений
Системный склероз
Классификация – клиническая и серологическая
Системный склероз (СС) является редким, но потенциально опасным для жизни состоянием. Существует три основных клинических подтипа, связанных с различными заболеваниями (): диффузный кожный (dc) SSc, характеризующийся широко распространенным и быстро прогрессирующим утолщением кожи (распространяющимся проксимальнее локтей и коленей), связанный с ранним поражением внутренних органов (легких, сердца и почек), ограниченный кожный ССД (lcSSc), характеризующийся ограниченным и непрогрессирующим утолщением кожи (ограничивается дистальными отделами конечностей), связанный с поздним заболеванием внутренних органов (легочная артериальная гипертензия, мальабсорбция), и перекрывающийся SSc, который может быть dcSSc или lcSSc с признаками другого заболевания соединительной ткани , такие как дерматомиозит или системная красная волчанка (СКВ) [9].Пациентов с CREST-синдромом (кальциноз, феномен Рейно, нарушение моторики пищевода, склеродактилия и телеангиэктазии) относят к ЛСКС. Некоторые пациенты с недифференцированным или смешанным заболеванием соединительной ткани могут иметь блестящую или полную кожу дистальных отделов пальцев, но у них должен быть основной критерий «уплотнение кожи или склероз проксимальнее пястной кости (MCP) или плюснефаланговой кости (MTP)». считается несовершеннолетним SSc (). Существуют дополнительные два малых критерия, необходимые для диагностики ССД у детей в соответствии с международным консенсусным соглашением () [10].В дополнение к степени утолщения кожи определенные профили аутоантител в сыворотке, связанные со склеродермией, помогают идентифицировать подмножества ССД и прогнозировать их поражение органов как у взрослых, так и у детей с началом ССД [3, 4, 6, 11, 12]. Например, антитело против топоизомеразы (Scl-70) можно ожидать у пациента с dcSSc, и это может вызывать беспокойство в связи с быстрым прогрессированием поражения кожи и развитием интерстициального заболевания легких (ИЗЛ). [3, 4, 6]. Четырьмя наиболее распространенными антителами при ССД у детей являются антитела к топоизомеразе (Scl-70) (20–34%), обычно связанные с dcSSc и ИЗЛ, как уже упоминалось, за которыми следуют антитела к центромерам (ACA) (2–16%). связанный с lcSSc и легочной артериальной гипертензией (ЛАГ), а также антитела к U1-RNP и Pm-Scl (полимиозит-склеродермия) (2–16%), оба из которых обычно обнаруживаются у пациентов с перекрывающимися синдромами SSc и другими соединительными заболевания тканей с более выраженными признаками артрита и миозита, такие как дерматомиозит или системная красная волчанка [3, 4, 6].Положительная доля пациентов с U1-RNP и Pm-Scl, вероятно, выше, чем сообщалось, поскольку некоторые авторы не включают случаи перекрывающегося SSc или смешанных заболеваний соединительной ткани в своих педиатрических когортах SSc. В целом, частота этих антител у детей отражает частоту клинической подгруппы dcSSc, lcSSc и перекрывающегося SSc [3, 4, 6]. По сравнению со взрослым SSc перекрывающийся SSc гораздо чаще встречается у детей (29% против 9%). , что отражается повышенной частотой антител U1-RNP и Pm-Scl и связано с более высоким процентом миозита и артрита, наблюдаемых у детей с ССД [3].Есть несколько других аутоантител, связанных с ССД, которые связаны с различными органными проявлениями; однако они гораздо реже встречаются при СД в детском возрасте. Одним из них является антитело к РНК-полимеразе III (POL3), которое относится к тяжелым почечным заболеваниям в форме склеродермического почечного криза. POL3 редко наблюдается при ССД в детском возрасте, но может быть обнаружен у 30% взрослых пациентов с ССД и отражает клинически значимую более высокую долю почечной недостаточности при ССД у взрослых (3, 6). Несмотря на то, что многие аутоантитела, связанные со склеродермией, были идентифицированы у взрослых ССД, существует высокая доля детей, 20-23%, которые имеют положительный результат АНА без идентифицированных специфических аутоантител [3, 6].
Типичная блестящая и толстая кожа рук с уплотнением кожи, распространяющимся вверх по предплечью (за пястно-фаланговые суставы) у пациента с системной склеродермией.
Таблица 1
Временные рекомендации по классификации Критерии несовершеннолетних системного склероза *
| Основным критерием (обязательно) | Устранение кожи / утолщение проксимальных к MCP или MTP-соединениям | ||
| Незначительный критерий (2 требуется) (Scleroderma конкретный орган участие органов) | |||
| COSCORE | |||
| Sclerodactyly | |||
| Мопиллярные изменения ногтей (мегакапилларии и авашелярные зоны) | |||
| Digital Tip lexers | |||
| Dysphasia | |||
| Gastroesophageal Reffull | |||
| Cardiac | |||
| Arrhhythmias | |||
| Сердце сбой | |||
| почек | |||
| почек | |||
| Scleroderma Renal Crisis | |||
| новая артериальная гипертония | |||
| легочной артериальной гипертензии (первичной или вторичной по отношению к ILD, оценивали по данным эхокардиографии) | |||
| Неврологические | |||
| невропатии | |||
| кистевой туннельный синдром | |||
| Musculoskeletal | |||
| фрикционные трется Сухожильные | |||
| Arthrititis | |||
| серологический | |||
| Антиатомное антитело | |||
| SSC-селективный автоантибоди es | |||
| Антитопоизомераза 1 (Scl-70), антицентромерная, антиРНК-полимераза I или III, анти-PM-Scl, антифибриллин |
Клинические проявления
Основные клинические проявления у детей SSc
Сосудистый
Общими признаками в начале детского SSc являются феномен Рейно (RP) (70%) и кожные изменения рук (60%), включая отек, склеродактилию и уплотнение проксимальнее MCP [4].В наибольшей изученной когорте педиатрических ССД (153 пациента) 53% имели как уплотнение кожи рук, так и РН, а 10% пациентов с РН также имели инфаркты пальцев [4]. На протяжении всего заболевания почти у всех будет РН (97%) и изменения кожи кистей (96%) [3].
Девочка-подросток с типичной (А) склеродактилией и (В) пальцевыми изъязвлениями и язвами.
Феномен Рейно — вазоспастическая реакция, приводящая к трехфазному изменению цвета рук (сначала бледность из-за вазоконстрикции, затем посинение вследствие цианоза и, наконец, краснота из-за реперфузии с последующим отеком и болью), связанная с ощущением онемения и покалывания .RP также может возникать в пальцах ног и других акральных областях, включая уши, кончик носа или язык. При ССД артериолы более узкие и жесткие из-за выраженной гиперплазии интимы и фиброза вследствие эндотелиальной дисфункции, что приводит к более выраженному РП с последующим повреждением/ишемией тканей [13]. Эти васкулопатические изменения идентифицируются в капиллярном ложе ногтевого ложа у этих пациентов с помощью капиллярной микроскопии, демонстрируя дилатацию, извилистость, кровоизлияние, выпадение (аваскуляризацию) и более позднюю разветвленность капилляров [14].Сообщается, что распространенность капиллярных изменений ногтевого ложа при ССД у детей составляет 50% [3, 4], но, вероятно, выше, если проводилась стандартизированная микроскопия. Плохая перфузия пальцев в конечном итоге приводит к изъязвлениям кончиков пальцев, изъязвлению (2), а в более тяжелых случаях — к аутоампутации. Здоровые люди также могут страдать феноменом Рейно без риска развития основного заболевания соединительной ткани (первичный синдром Рейно). У этих людей обычно никогда не развиваются язвы на кончиках пальцев или капиллярные изменения ногтевого валика, поскольку у них нет основной васкулопатии.Признаки, которые помогают клиницисту дифференцировать первичный феномен Рейно от феномена, вторичного по отношению к заболеванию соединительной ткани (ЗСТ), как у детей, так и у взрослых, следующие: отсутствие изменений капилляров ногтевого валика, изъязвление и/или изъязвление кончиков пальцев и отрицательный АНА [15, 16].
;;9 и 2,8 года между первыми признаками заболевания и диагнозом [3, 4]. В начале клинического течения кожа отечна, особенно на дистальных отделах конечностей; редко присутствует поражение проксимальных отделов конечностей, лица и туловища. Фаза индурации, в честь которой названа склеродермия, характеризуется потерей естественной податливости кожи и наличием пальпируемой толщины кожи. Кожа приобретает блестящий, напряженный вид с сужением дистальных отделов пальцев (и ). Со временем, по мере того как кожа утолщается, а нижележащие структуры, такие как сухожилия, поражаются и укорачиваются, суставы пальцев начинают терять диапазон движений как при разгибании, так и при сгибании, а в более тяжелых случаях возникает деформация «когтеобразной руки», что сильно влияет на работоспособность. повседневная деятельность (ADL).Типичное лицо склеродермии с натянутой кожей и атрофией кожи характеризуется сморщенным носом, тонкими сжатыми губами, маленьким ртом, выступающими зубами и невыразительным видом (4). Толщина кожи оценивается как легкая, умеренная и тяжелая по всему телу, и кумулятивный балл получается с использованием модифицированной шкалы кожи Роднана (mRSS) [17]. mRSS получают в ходе лонгитюдных посещений, и можно рассчитать скорость прогрессирования толщины кожи (STPR), что помогает прогнозировать время поражения внутренних органов у пациентов с dcSSc.Высокий STPR связан со склеродермическим почечным кризом [18].
Невыраженное лицо при склеродермии. Девочка препубертатного возраста с уменьшенным ротовым отверстием и «поджатыми губами».
Другие кожные проявления при СС включают подкожные отложения кальция (кальциноз кожи), которые могут возникать в точках давления, обычно обнаруживаемых на разгибательных поверхностях суставов кистей при СС, и могут иногда выдавливаться через кожу подобно дерматомиозиту. Эти поражения могут быть болезненными и изъязвляться.Однако они чаще связаны с хроническим заболеванием и редко присутствуют во время начала заболевания и постановки диагноза. Телеангиэктазии также являются проявлением ССД, чаще всего встречаются в варианте lcSSc (ранее известном как CREST-синдром) и обычно локализуются на лице и верхних конечностях.
Другие органные проявления при ССД (в порядке убывания частоты) включают желудочно-кишечные, легочные, скелетно-мышечные, сердечные, почечные и неврологические симптомы. Желудочно-кишечные симптомы встречаются примерно у половины детей, хотя более детальное исследование часто указывает на наличие аномалий у большего процента [3].Нарушение моторики пищевода и гастроэзофагеальный рефлюкс часто приводят к дисфагии и эзофагиту. Дистальная дисфагия, особенно при приеме твердой пищи, вызывающая ощущение «застревания пищи» в середине грудной клетки, является типичной и представляет собой дисфункцию дистальной гладкой мускулатуры пищевода. Гастропарез с замедленным опорожнением желудка в дополнение к дисфункции пищевода может привести к усилению симптомов рефлюкса. Обычно пациенты жалуются на ночной кашель в положении лежа на животе из-за тихой аспирации и солоноватый привкус во рту при пробуждении из-за тихого рефлюкса.Хронический рефлюкс может привести к стриктурам пищевода. Оценка желудочно-кишечного тракта с использованием манометрии и внутрипищеводного 24-часового рН-мониторинга считается чувствительным индикатором сниженного тонуса пищевода и рефлюкса [19]. Однако в педиатрии эти методы используются реже. Вместо этого обычно используется исследование глотания для оценки аспирации и нарушений моторики с последующим исследованием верхних отделов желудочно-кишечного тракта с последующим исследованием тонкой кишки. Если поражена тонкая кишка, спазмы, диарея и запор могут быть результатом перистальтической дисфункции.Эпизоды псевдообструкции с постпрандиальным вздутием живота, болью и тошнотой могут возникать из-за функциональной кишечной непроходимости. Могут наблюдаться избыточный бактериальный рост, стеаторея, потеря веса, заворот и даже перфорация. Заболевание толстой кишки проявляется в виде широкоротых дивертикулов и потери нормальной архитектуры толстой кишки. Другим осложнением поражения ЖКТ при ССД является острое желудочно-кишечное кровотечение, связанное с эктазией антральных вен желудка (ГАВЭ), требующее фотокоагуляции. Это состояние редко встречается у детей и связано с ранним dc SSc, РНК-полимераза III положительных пациентов [20].
Поражение легких при педиатрическом ССД составляет в литературе от 30 до 70% и включает интерстициальное заболевание легких (ИЗЛ), легочную артериальную гипертензию (ЛАГ) (либо первичную, которая возникает в результате васкулопатии, либо вторичную из-за ИЗЛ), и аномальные легочные функциональные тесты (PFTs). Особое беспокойство вызывают аномалии PFT, такие как снижение форсированной жизненной емкости легких (ФЖЕЛ) и диффузионной способности монооксида углерода (DLCO) [21]. Интерстициальное заболевание легких (ИЗЛ) обычно начинается с воспалительной фазы, авеолита, характеризующегося смешанным клеточным инфильтратом в интерстиции легкого, который распространяется в альвеолярные пространства.Бронхоавеолярный лаваж (БАЛ) демонстрирует увеличение нейтрофилов и/или эозинофилов в дополнение к альвеолярным макрофагам [22]. На наличие авеолита также указывают затемнения по типу матового стекла на компьютерной томографии высокого разрешения (КТВР) легких. По мере прогрессирования ИЗЛ воспаление стихает, так как происходит переход в сторону фиброза с утолщением альвеолярных стенок и ремоделированием легкого. Прогрессирующий легочный фиброз может привести к рестриктивному заболеванию легких от умеренной до тяжелой степени со значительным снижением ФЖЕЛ и DLCO.Клинически ИЗЛ обычно проявляется в виде медленно прогрессирующей одышки при физической нагрузке в течение многих лет. Для сравнения, ЛАГ проявляется быстрым прогрессированием одышки при физической нагрузке в течение нескольких месяцев. В условиях ЛАГ PFT демонстрируют снижение DLCO и аномальные результаты эхокардиограммы с предполагаемым систолическим давлением в легочной артерии (ЛА) или правом желудочке (ПЖ)> 40 мм рт.ст., что подтверждается катетеризацией правых отделов сердца со средним давлением ЛА> 25 мм рт.ст. Для большинства детей с ССД или подозрением на ССД первоначальная оценка включает рентгенографию грудной клетки, тесты функции легких, включая DLCO, и эхокардиограмму.Дополнительные сердечно-легочные тесты проводятся, если обнаруживаются аномалии или пациент слишком молод, чтобы надлежащим образом завершить тестирование (например, PFT). Аномальные PFT требуют оценки с помощью HRCT грудной клетки и, возможно, BAL, если требуются дополнительные доказательства воспаления (авеолит) или необходима оценка инфекции перед началом терапии [21, 23]. Признаки ЛАГ на эхокардиограмме более формально оцениваются при катетеризации правых отделов сердца.
По сравнению с началом ССД у взрослых поражение опорно-двигательного аппарата чаще встречается у детей с ССД.Приблизительно 30–40% страдают воспалительным артритом (суставные выпоты) в дополнение к типичному «сухому синовиту» склеродермии, выражающемуся в фиброзе сухожилий, пересекающих суставы, что ограничивает диапазон их движений (ROM). В начале патологического процесса, особенно у пациентов с dcSSc, теносиновит/бурсит вызывает ощутимый шум трения сухожилий («кожистый хруст») при разгибании или сгибании сустава [24]. При миопатии у больного обычно отмечается симметричная проксимальная слабость, особенно плечевого пояса и плечевых мышц, иногда с выраженной атрофией.Миозит также проявляется повышением уровня мышечных ферментов и изменениями групп мышц на МРТ, сходными с таковыми при ювенильном дерматомиозите (ЮДМ). Биопсия мышц при ССД отличается от таковой при ЮДМ наличием большего количества фиброза и наличием утолщенных капилляров [25]. Дети с dcSSc и миозитом особенно подвержены риску тяжелого поражения сердца, включая дефицит перфузии миокарда и дилатационную кардиомиопатию [26].
Поражение сердца , хотя и редко, но является основной причиной связанной со склеродермией смертности у детей с ССД, о чем свидетельствуют 10 из 15 смертей в когорте Martini et al. [4] и 5 из 32 смертей в Scalapino et al. когорта [3].Сердечные проявления являются результатом сочетания миокардиального фиброза, сосудистой недостаточности и воспаления и включают сердечную блокаду, аритмию, застойную сердечную недостаточность, кардиомиопатию и перикардит. В отличие от детского ССД, смертность от склеродермии у взрослых в первую очередь обусловлена поражением легких, как ИЗЛ, так и ЛАГ [27].
Легкая почечная дисфункция не редкость при ССД и является вторичной по отношению к васкулопатии, а не к гломерулонефриту, который наблюдается при системной красной волчанке.Это подтверждается патологическими данными пролиферации интимы междольковых артерий почек и отсутствием активного мочевого осадка (т.е. эритроцитарных цилиндров). Напротив, более тяжелая и болезненная форма заболевания почек, склеродермия , почечный криз (SRC), встречается только примерно у 15% взрослых пациентов с ССД и еще реже у детей. СРК характеризуется ускоренной артериальной гипертензией, почечной недостаточностью, микроангиопатической гемолитической анемией и тромбоцитопенией.При SSc у взрослых SRC ранее имел 20% выживаемость в течение 1 года, но теперь выживаемость составляет 80% из-за появления ингибиторов ангиотензинпревращающего фермента (ACEI) [27]. SRC чаще всего наблюдается на ранних стадиях заболевания у пациентов с РНК-полимеразой III dcSSc и, как полагают, усиливается при более высоких дозах кортикостероидов. Почечный криз у детей с ССД встречается редко, с менее чем 5% случаев, и только 5 случаев зарегистрированы (2 из которых умерли от почечной недостаточности) среди трех больших опубликованных когорт [3, 4, 28].
Исход
Несмотря на все возможные поражения органов при ССД у детей, она имеет гораздо более благоприятный прогноз через 5, 10 и 15 лет по сравнению со ССД у взрослых с более низкой частотой тяжелого поражения органов. Foeldevari и коллеги [28] недавно сообщили, что показатели выживаемости Kaplan Meier в течение 10 лет для детей составляют 98% по сравнению с 75% у взрослых. Лечение ССД зависит от поражения органов, но общие меры, такие как антациды для защиты ЖКТ, прокинетики для нарушения моторики, чередование антибиотиков для мальабсорбции, а также сосудорасширяющие средства при феномене Рейно (RP) (могут также помочь при ЛАГ), нестероидные Обычно рекомендуются противовоспалительные препараты (НПВП) и физиотерапия (ЛТ) при артрите или тендините.Кортикостероиды при миозите, артрите и других воспалительных клинических проявлениях следует использовать с осторожностью, поскольку они могут спровоцировать склеродермический почечный криз. Более конкретные рекомендации по лечению различных органных проявлений были согласованы на консенсусной конференции международных экспертов по склеродермии и опубликованы в 2009 г. [29]. Примечательно, что в лечении раннего диффузного кожного заболевания как у детей, так и у взрослых произошел переход от использования D-пеницилламина к микофенолата мофетилу (ММФ) из-за потенциальных антипролиферативных эффектов, хотя это не было включено в рекомендации по лечению, которые обсуждались ранее. 30].
Локализованная склеродермия
Классификация
Локализованная склеродермия, также известная как «морфея», имеет другой тип поражения кожи, чем системный склероз (СС), и включает несколько подтипов, классифицированных по глубине и характеру поражения(й). Группа заболеваний характеризуется фиброзом, который в основном ограничивается кожей и подкожной клетчаткой; однако более глубокие формы также включают фасции, мышцы, сухожилия и суставную капсулу. Традиционная классификация, предложенная Peterson et al.[31] описали пять подклассов, включая бляшечную морфею, генерализованную морфею, буллезную морфею, линейную морфею, глубокую и пансклеротическую морфею (1). Однако совсем недавно консенсус экспертов внес изменения, чтобы предоставить более клинически применимые классификации, названные «Критерии Падуи», которые включают подтип «смешанного морфеа», учитывая, что распространенность этой категории составляет 15% () [32].
Таблица 2
| A: LS ° Подтипы: Mayo Классификация ‡ |
|---|
| Налет Morphea † |
| Morphea ан бляшек † |
| каплевидный Morphea |
| атрофодермия из Pasini и Пьерини |
| Обобщенная Morphea † |
| Буллезная Morphea |
| линейный Morphea † |
| линейная Morphea (линейная склеродермия) † |
| En купе де сабля † |
| Progressive гемифациальный атрофия |
| Deep Morphea † |
| Подкожный Morphea † |
| эозинофильный фасциит | Morphea profunda † |
| Отключения pansclerotic Morphea детей |
| б: Предлагаемые подтипов для ювенильного LS °: Падуя Предварительной Классификации ‡ | |||
|---|---|---|---|
| Описанного Morphea | Овальные/круглые поражения
| 1||
| Linear Scleroderma | Линейные поражения могут включать дерма, подкожная клетчатка, мышцы, кости
| Обобщенные Morphea | ≥ 4 Большие бляшки (> 3см) по крайней мере 2 7 анатомических областей: голова/шея, правая верхняя конечность, LUE, RLE, LLE, передняя часть туловища, задняя часть туловища | |
| Пансклеротическая морфеа | Окружное поражение конечности (за исключением кончиков пальцев рук и ног) Все глубины кожи/подкожной клетчатки /мышца/кость | ||
| Смешанная форма морфеа | Комбинация двух или более вышеперечисленных подтипов | ||
Активность заболевания и признаки повреждения (клинические и гистологические данные, клинические и гистологические признаки) Сходны с СЛ
более ранняя более активная фаза заболевания и более поздняя более фиброзная фаза ().Ранние активные поражения при СЛ характеризуются фиолетовой воспалительной каймой или «сиреневым кольцом» (1) и индурацией кожи по всему очагу поражения, включая границу. Во время этого активного состояния поражения также могут увеличиваться, и могут накапливаться новые поражения [33]. Гистологическая картина на ранних стадиях заболевания состоит из периваскулярного инфильтрата преимущественно из лимфоцитов с примесью редких плазматических клеток и эозинофилов в глубоком ретикулярном слое дермы и подкожной клетчатке, сопровождающемся утолщением коллагеновых пучков, уменьшением эластических волокон и набуханием эндотелиальных клеток [34, 35].С течением времени поражение болезнью накапливается и проявляется увеличением толщины кожи, особенно в центре поражения, иногда оставляя склеротический очаг цвета слоновой кости (). Гистологическая оценка кожи выявляет утолщенный гипоцеллюлярный (гомогенизированный) коллаген, который заменил прежний воспалительный инфильтрат вокруг придатков дермы, оставив после себя атрофические эккринные железы и волосяные фолликулы. Кроме того, уменьшается количество капилляров с фиброзной стенкой и суженным просветом [34, 35].Клинический результат этих находок отражается в кожной и подкожной атрофии. При физикальном осмотре, когда дерма атрофична, наблюдается отсутствие роста волос в месте поражения, видимые вены и легкий вид «обрыва» на коже (). Подкожная атрофия существует в непрерывном диапазоне и может быть как легкой, как уплощение кожи, так и более «выемчатым» вогнутым видом жировой ткани при более умеренных поражениях, а в самых тяжелых случаях до такой степени, что можно увидеть мышцы, движущиеся под кожей.Поствоспалительная гипер- и гипопигментация также являются результатом предшествующего воспаления и часто являются признаками, которые привлекают внимание врача к поражению. Часто семьи пропускают раннюю активную фазу и считают эритему/фиолетовый цвет скорее синяком или травмой, но именно тот факт, что «синяк, который не проходит» (поствоспалительная гиперпигментация), часто побуждает семью искать медицинская помощь ().
Фазы локализованной склеродермии и связанные с ней признаки.Признаки активного заболевания, эритема, уплотнение/отек кожи и новые или увеличивающиеся очаги поражения возникают первыми и уступают место признакам поражения заболеванием, диспигментации, атрофии и утолщению кожи.
Типичное «сиреневое кольцо», наблюдаемое на границе множественных бляшек на туловище, указывающее на активное заболевание, любезно предоставлено Еленой Поуп из Children’s Sick Kids, Торонто.
Восковидно-желтый очаг поражения в центре бляшки, остающийся на правой стороне шеи спустя годы после начала локализованной склеродермии.
Кожная атрофия LS, проявляющаяся в виде «капельных» поражений у девочки-подростка с генерализованной морфеей.
Патогенез (общий для ССД и ССД)
Общие гистологические данные на коже пациентов с ССД и СЛ иллюстрируют их общую патофизиологию, несмотря на различные клинические особенности заболевания. Оба являются аутоиммунными заболеваниями с воспалительным, фиброзным и сосудистым компонентом. Популяции мононуклеарных клеток, идентифицированные как в образцах кожи SSc, так и в LS, представляют собой преимущественно Т-клетки, как CD8, так и CD4-положительные, с более высоким процентом клеток CD4 (T-хелперы) [36, 37]. Ассоциированные с Th-клетками цитокины и хемокины, такие как интерлейкины [IL]-2, -4, -6, -8, -13 и TNF-a, были идентифицированы в циркулирующей сыворотке и стимулированных PBMC у пациентов с LS и SSc [38–38]. 43].Следует отметить, что некоторые сывороточные цитокины были обнаружены в более высоких концентрациях у пациентов с LS по сравнению с СС, что интересно, поскольку LS называют «локализованным заболеванием»; однако это может подтверждать высокую частоту внекожных проявлений (примерно 25%), продемонстрированную при СЛ [44]. Было выявлено несколько корреляций между клиническими переменными тяжести заболевания LS и цитокинами и их растворимыми рецепторами, такими как количество бляшек и линейных поражений (IL-13, TNF-a, sIL-2r, sIL-6r), генерализованная морфия и субпопуляции линейной склеродермии (IL-2,-4,-6,-8,-13, TNF-a, sIL-2r, sIL-6r) и корреляция антител, антигистоновые и/или оц-ДНК антитела (IL -4, -6, TNF-a, sIL-2r, sIL-6r) [38–40, 45–47].Эти ассоциированные с Т-клетками цитокины, в свою очередь, дополнительно стимулируют фибробласты и эндотелиальные клетки к выработке трансформирующего фактора роста бета (TGF-β) и фактора роста соединительной ткани (CTGF), которые считаются основными стимуляторами тканевого фиброза (за счет увеличения коллагена). продукция) и повреждение эндотелиальных клеток (через влияние молекул адгезии ICAM-1 и VCAM-1) [48–51].
Микрохимеризм является еще одной интересной областью, изучаемой в отношении общего патогенеза LS и SSc.Клинически и гистологически LS и SSc имеют общие характеристики с хронической болезнью «трансплантат против хозяина» (cGVHD) (1), типом микрохимеризма, который играет роль в патогенезе. Химерные клетки были идентифицированы у пациентов с LS и SSc с использованием методов ПЦР в реальном времени [52, 53].
Линейное поражение туловища (A) и ног (B) у ребенка раннего возраста со склеродерматозной хронической болезнью «трансплантат против хозяина».
Аутоиммунитет, как один из компонентов распространения LS, подтверждается профилем аутоантител, сопутствующими сопутствующими аутоиммунными заболеваниями и семейным анамнезом аутоиммунных заболеваний, наблюдаемых у пациентов с LS.Аутоантитела обычно положительны при LS, включая антиядерные антитела (ANA), антигистоновые антитела (AHA) и антитела к одноцепочечной ДНК (ss-DNA Ab) [54]. ANA является классическим серологическим маркером аутоиммунного заболевания и обнаруживается у пациентов с СЛ с высокой частотой, в диапазоне от 42 до 73% [5, 55]. Считается, что более высокие титры связаны с ранним развитием заболевания [55] и повышенным риском внекожных проявлений [44, 56]. Распространенность оц-ДНК при СЛ составляет примерно 50% [57] и связана с линейной склеродермией (особенно с контрактурами суставов), вовлечением глубоких мышц и увеличением количества анатомических участков; все это отражает увеличение тяжести заболевания [56, 58].Было показано, что в подгруппе пациентов с LS одноцепочечное антитело к ДНК отражает статус активности заболевания, при этом титры снижаются по мере клинического улучшения кожных поражений [54, 56]. АГА встречается с аналогичной частотой при СЛ (47%) [59] и также коррелирует с показателями тяжести заболевания; включая общую площадь вовлеченной поверхности тела, количество поражений, линейное заболевание и наличие контрактур суставов [54, 56]. В исследовании, посвященном линейной склеродермии, сочетание положительных ss-ДНК и AHA было обнаружено у 23% пациентов (взрослых и детей с СЛ) и достоверно коррелировало с наличием контрактур суставов [56].Вышеупомянутые антитела указывают на тяжесть заболевания и могут помочь в стратификации пациентов с ранним диагностированным заболеванием, предполагая возможное начало более агрессивной терапии и усиленное наблюдение.
Сопутствующие и семейные ревматические и аутоиммунные заболевания более распространены при СЛ (детском и взрослом) по сравнению с общей популяцией [60]. Сопутствующее заболевание было зарегистрировано у 5–10% детей [7, 44, 60] и 30% взрослых с СЛ [60]. Наиболее распространенными заболеваниями были дерматологические аутоиммунные заболевания (псориаз, витилиго, очаговая алопеция), за которыми следовали ревматические состояния (ювенильный идиопатический артрит, ревматоидный артрит (РА), системная красная волчанка (СКВ) и синдром Шегрена) [7, 60].При оценке по подтипу СЛ частота сопутствующего аутоиммунного заболевания была значительно выше у пациентов с генерализованным морфеозом [60]. Семейные ревматические заболевания и другие аутоиммунные заболевания были зарегистрированы у 12-25% всех пациентов (как у детей, так и у взрослых), наиболее частым из которых был псориаз, за которым следовали РА, СКВ, ССД, СЛ, рассеянный склероз и тиреоидит [5, 7]. , 60]. Вновь была продемонстрирована связь между генерализованной морфеей и повышенной частотой этих заболеваний [5].
Клинические проявления
Хотя термин «локализованная склеродермия» или «морфея» объединяет все подтипы в одну категорию, их кожные/подкожные проявления и внекожные ассоциации совершенно уникальны. Линейная склеродермия (LiScl) является наиболее распространенным подтипом, встречающимся у 50–60% детей с LS, и характеризуется линейной полосой или полосой, поражающей дерму, подкожную клетчатку и часто подлежащие мышцы, сухожилия и кости. LiScl обычно возникает в виде одиночной линейной односторонней полосы на конечностях, туловище или голове (5, 7).Наиболее часто поражаются ноги, за ними следуют руки, лобная часть головы и туловище. Поражения LiScl, поражающие конечности, вызывают наибольшее беспокойство, когда они пересекают сустав, поскольку существует вероятность склероза и уплотнения нижележащих мышц, сухожилий и суставной капсулы, что приводит к контрактуре сустава и связанной с этим дисфункции конечностей. Особое беспокойство у детей с LiScl вызывает поражение эпифизарной зоны роста, что может привести к необратимому укорочению или атрофии конечности.Ортопедические осложнения регистрируются примерно у 30–50% пациентов с LiScl конечностей (конечностей) [7, 58, 61]. Peterson и соавторы сообщили о легкой или умеренной инвалидности при последующем наблюдении у пациентов с LiScl конечностей в их когорте (среднее время наблюдения 9,2 года) [1].
Линейная склеродермия, затрагивающая одностороннее поражение правой верхней конечности (А). У этой молодой женщины ассоциированные контрактуры лучезапястных, пястных и фаланговых суставов. Суставные контрактуры кисти отчетливо видны по сравнению с контралатеральной стороной при активном разгибании (В) и сгибании (С).
Приблизительно у 25% пациентов с LiScl наблюдается поражение волосистой части головы [5]. Линейные поражения кожи головы и лба называются «удар саблей» (ECDS), потому что линейная депрессия напоминает шрам от саблевидной раны. Прогрессирующая гемифациальная атрофия, также известная как синдром Парри-Ромберга (PRS), представляет собой еще один линейный вариант LS, вызывающий гемиатрофию лица, включая нижнюю, верхнюю челюсть, язык, подкожную и мышечную ткани (4). Как правило, ECDS ассоциируется с кожными проявлениями диспигментации и толщины кожи, тогда как PRS не является и обычно имеет более выраженную подкожную атрофию.Эти расстройства могут возникать вместе, либо в одном и том же месте (сторона лица), либо на противоположных сторонах [62]. Несмотря на противоречивость, ECDS и PRS, вероятно, представляют собой разные спектры одного и того же состояния, что подтверждается сходными проявлениями и преобладанием неврологической симптоматики [62, 63] и клеточных инфильтратов (лимфоцитарный инфильтрат сосудисто-нервных пучков) кожи [64] и ткани головного мозга [65]. ]. PRS и ECDS несут одинаковый риск неврологического поражения, при этом у 8–20% пациентов с этими состояниями наблюдается поражение центральной нервной системы, выражающееся в судорогах, хронических головных болях, неврите зрительного нерва и, реже, в нервно-психических изменениях или ишемическом инсульте [62, 63]. ].Также сообщалось об ортодонтических и офтальмологических аномалиях с одинаковой частотой при обоих состояниях [63]. Если начало заболевания (ECDS или PRS) происходит в возрасте до 10 лет, существует особый риск скелетной гипоплазии пораженной стороны лица из-за незрелости скелета, что приводит к стоматологическим и косметическим аномалиям [66]. ].
Саблезубое поражение (ECDS), поражающее волосистую часть черепа и голову односторонним образом (A), вызывающее умеренную потерю подкожной ткани и деформацию скелета (B).
Тяжелый пример синдрома Парри-Ромберга/гемифациальная атрофия правого лица.
Поверхностная ограниченная морфия, ранее называемая бляшечной морфеей по классификации Петерсона, является наиболее частым проявлением СЛ у взрослых (60%) и составляет примерно одну четверть СЛ у детей [5, 60]. Он характеризуется одной или несколькими овальными или округлыми участками уплотнения с восковым центром цвета слоновой кости и фиолетовым ореолом по периферии диаметром от 1 до 30 см. Эти поражения обычно возникают на туловище или проксимальных отделах конечностей (4).Поверхностный вариант ограничен эпидермисом и дермой. Глубокая ограниченная морфеа, ранее называемая подкожной морфеей, глубокой морфеей или глубокой морфеей, представляет собой круглое/овальное поражение, расположенное глубже в ткани. Эти поражения поражают дерму и подкожную клетчатку с различным проникновением в фасции и мышцы. Как правило, имеются только тонкие кожные изменения (такие как небольшая эритема и истончение дермы), но присутствует большое количество воспалительных инфильтратов в глубоких подкожных тканях и фасциях, вызывающих депрессию в жировой ткани («выемчатый» вид) и утолщение. , стянутая кожа.
Типичное изолированное овальное поражение на туловище, соответствующее поверхностным очерченным морфеям.
Генерализованная морфеа (ГМ) определяется наличием более 3 бляшек, каждое из которых имеет ширину более 3 см. Обычно бляшки сливаются и поражают несколько анатомических областей, чаще всего на туловище. ГМ — редкий подтип, встречающийся менее чем у 10% пациентов с СЛ [5, 7]. Как упоминалось ранее, этот подтип часто связан с несколькими аутоиммунными явлениями, включая антитела к одноцепочечной ДНК и AHA, а также сопутствующий и семейный анамнез аутоиммунных заболеваний.Системные симптомы, такие как миалгия, артралгия и утомляемость, чаще встречаются при ГМ [7, 60].
Пансклеротическая морфеа — редкая разновидность СЛ, поражающая только 1–2% пациентов и обычно приводящая к инвалидности. Первоначально он поражает конечности, но позже может мигрировать на туловище, не затрагивая только лицо и дистальные области пальцев рук и ног (4). Отмечается быстро прогрессирующий фиброз глубоких слоев дермы, подкожной клетчатки, фасций и мышц, иногда вовлекающий кости. Это приводит к значительной контрактуре суставов, атрофии мышц, изъязвлениям кожи и иногда к рестриктивной дыхательной недостаточности, если задействован грузовик.Развитие плоскоклеточного рака является дополнительным осложнением при пансклеротической морфее, особенно в области хронических ран [67].
Пансклеротическая морфеа, поражающая конечности по окружности и не затрагивающая дистальные отделы пальцев ног и пальцев.
Смешанная форма морфеа является последним подтипом, рассмотренным на основе консенсуса Падуи, и определяется наличием более чем одного клинического подтипа у одного пациента с СЛ. Смешанная форма морфеа довольно распространена и наблюдается примерно у 15% детей с СЛ.Наиболее частым проявлением смешанного морфеа является сочетание линейного и бляшечного морфеа [5].
Буллезный морфеоз иногда считают редким подтипом СЛ. Тем не менее, многие эксперты согласны с тем, что буллезные поражения являются вторичными по отношению к агрессивному линейному или глубокому СЛ и, вероятно, отражают нарушение лимфатического оттока. Эозинофильный фасциит (ЭФ), когда-то считавшийся типом глубокого морфеа по критериям Петерсона, не считается подтипом СЛ по критериям Падуи. Он часто ведет себя как смесь глубокой морфеи и линейной склеродермии, что приводит к глубокой инфильтрации и контрактурам суставов [31].)
Внекожные проявления
Внекожные проявления (ВКМ) нередки при СЛ и возникают примерно у четверти пациентов на протяжении всего заболевания [44]. Согласно большому международному исследованию 750 детей с LS, внеклеточное маточное окошко чаще встречается у пациентов с LiScl и в основном состоит из ортопедических осложнений (19% в целом в группе, 50% у детей с LiScl конечностей), неврологических или глазных нарушений (5% всей группе, 40% у лиц с LiScl головы), феномен Рейно (2.1% всей группы) и другие аутоиммунные состояния, такие как тиреоидит (2% всех подтипов) [44]. Менее распространенными ЭКМ являются желудочно-кишечные, респираторные и почечные, все они встречаются менее чем у 2% из 750 обследованных пациентов [44]. Эти оценки могут быть ниже, чем истинный процент ECM, поскольку в анализ были включены только пациенты с симптомами. Другим ограничением исследования было отсутствие контрольной популяции для оценки распространенности этих проявлений в сопоставимой детской популяции.В другом исследовании, в котором изучались желудочно-кишечные проявления у пациентов с СЛ (такие как гастроэпофагеальный рефлюкс) и изменения неврологической визуализации у пациентов с ECDS/PRS, исследователи обнаружили более высокую частоту внеклеточного матрикса; 20% и 60% соответственно [63, 68]. Интересно, что в исследовании Zulian et al., включавшем 750 детей с LS, из 168 пациентов с внеклеточным матриксом у 25% были суставные, неврологические и глазные проявления, не связанные с местом поражения кожи, т.е. артрит периферического сустава с непораженной вышележащей кожей и подкожной клетчаткой. , что еще раз подтверждает гипотезу о том, что «локализованная склеродермия» является более системным процессом [44].У 24 из 750 детей (3,2%) было выраженное поражение глаз, у большинства из которых была линейная склеродермия головы (67%) [69]. Эти результаты подтверждают рутинный офтальмологический скрининг пациентов с LS на наличие признаков воспаления глаз, особенно у пациентов с линейной склеродермией головы. Этот безопасный инструмент скрининга может выявить клинически «тихую» болезнь, а лечение может предотвратить долгосрочные последствия для зрения.
Оценка
Ни один лабораторный тест не является диагностическим для LS. Несмотря на «локализованное» заболевание, было выявлено несколько аутоантител, связанных с LS, которые могут помочь в диагностике и указать на более тяжелое/вовлеченное заболевание (, см. раздел об аутоиммунитете ).В частности, положительность ANA, AHA и одноцепочечной ДНК обнаруживалась с относительно высокой частотой (40–50%). Кроме того, ревматоидный фактор присутствует у трети больных, особенно у больных ГМ, а также коррелирует с тяжестью заболевания (количеством поражений) [5]. Общие маркеры воспаления, включая скорость оседания эритроцитов и уровень сывороточного иммуноглобулина, могут быть полезными маркерами активности заболевания у отдельных пациентов с СЛ, особенно с глубокими или эозинофильно-подобными вариантами.Однако у большинства пациентов с активными поражениями LS эти маркеры не повышены. Диагноз LS обычно ставится при клиническом обследовании, и, если есть сомнения, биопсия кожи и подкожной клетчатки может помочь в диагностике. Дифференциальная диагностика ограничена и включает два состояния кожи с хорошо выраженными бляшечными поражениями: склерозирующий и атрофический лихен и атрофодермию Пазини и Пьерини. Оба являются поверхностными дермальными и эпидермальными воспалительными состояниями, которые, по мнению некоторых, относятся к более мягкому спектру локализованной склеродермии.В дифференциальный диагноз включают кожную Т-клеточную лимфому, особенно с более глубоким багровым изменением цвета кожи, и коллагеному, обычно проявляющуюся уплотнением кожи и подкожного слоя без пигментных изменений с рождения или в раннем детстве. Другие возможные состояния связаны с определенным воздействием и, как правило, имеют более широко распространенные поражения, такие как реакция «трансплантат против хозяина» (состояние после трансплантации), нефрогенная фиброзирующая дерматопатия (гадолиний вводят при нарушении функции почек) и поздняя кожная порфирия (воздействие солнца).
Для оценки поражений кожи и подкожной клетчатки при LS используется несколько инструментов, таких как клиническая оценка кожи, инструменты измерения и визуализация.Клинические оценки кожи обычно используются врачами и включают модифицированную шкалу Роднана [17] и инструмент оценки локализованной склеродермии кожи (LoSCAT), который объединяет параметры активности заболевания и повреждения [33, 70]. Компьютеризированная оценка кожи (CSS) может использоваться для объективного определения размера поражения [71]. Инструменты измерения, в том числе дюрометр, который измеряет степень твердости кожи [72], и кутометр, который измеряет эластичность кожи [73], также были предложены в качестве полезных.Кроме того, для диагностики или оценки можно использовать несколько типов визуализации, таких как термография [74, 75], лазерная допплеровская флоуметрия [76], УЗИ [77, 78] и МРТ [79]. Некоторые из этих методов были проверены в LS, включая LoSCAT, CSS, ультразвук с использованием более высоких частот (10–25 МГц) и МРТ. Однако в настоящее время стандартные меры широко не используются, и оценка зависит от методов, специфичных для центра, которые зависят от опыта исследователя.
Недавние усилия Альянса по исследованиям детского артрита и ревматологии (CARRA) были сосредоточены на совместных проектах исследователей детской ревматологии и дерматологии в Северной Америке.Их цель состоит в том, чтобы установить конкретные клинические меры для оценки активности и повреждения, которые помогут в мониторинге прогрессирования и состояния болезни при LS. Эти проекты находятся в процессе оценки клинических показателей, таких как LoSCAT, и определения других с помощью многоцентровых исследований с группой клинических и ультразвуковых исследований локализованной склеродермии (LOCUS). Группа LOCUS состоит из экспертов по СЛ, как детских ревматологов, так и дерматологов, которым было предложено ранжировать клинические переменные, наиболее сильно отражающие активность заболевания и повреждения при СЛ.Консенсусное согласие >75% и высокая содержательная достоверность (модифицированная каппа-статистика 0,88–1,00) в отношении четырех параметров активности; появление новых поражений, расширение существующих поражений, эритематозный/фиолетовый оттенок на границе и уплотнение кожи на границе поражения [33]. Параметры повреждения кожных заболеваний, получившие наивысшее место среди группы, включали поствоспалительную гиперпигментацию и гипопигментацию, кожную и подкожную атрофию (согласие 88% и межпунктовый индекс достоверности 0.88 – 1,0) [70].
После начала системной иммуносупрессивной терапии у пациентов с умеренным и тяжелым СЛ признаки активности заболевания обычно исчезают в течение нескольких месяцев, а некоторые признаки повреждения уменьшаются от легкой до умеренной степени [80–85]. Именно после этого начального улучшения важно выявление субклинического заболевания, чтобы не отменять иммуносупрессию слишком рано, что может привести к рецидиву [80, 83, 84]. Недавнее исследование педиатрического СЛ выявило 28% рецидивов после средней продолжительности лечения метотрексатом, равной 2.4 года. Было обнаружено, что линейный подтип конечностей и пожилой возраст в начале заболевания являются значимыми предикторами рецидива, и, хотя это не значимо, была заметная разница в продолжительности лечения метотрексатом между рецидивирующими и нерецидивирующими пациентами [85]. Тем не менее, по-прежнему трудно обнаружить субклиническое заболевание, поэтому используются различные методы визуализации и тепловизионного исследования.
Лечение
Подход к лечению СЛ изначально зависит от тяжести заболевания, которое характеризуется подтипом СЛ, возможным вовлечением более глубоких тканей и структур (таких как суставы), локализацией СЛ и степенью активности заболевания.Продолжительность заболевания действительно влияет на состояние активности болезни, при этом большинство поверхностных ограниченных или бляшечных поражений «выгорают» через три-пять лет. Любой оставшийся центральный склероз может со временем смягчиться без вмешательства. Если остаются только параметры поражения болезнью и нет расширения, эритемы или других клинических параметров активности заболевания, допустимо простое наблюдение за поражением. Однако, если есть какие-либо признаки активного заболевания, как детские ревматологи, так и дерматологи согласны с тем, что одно поверхностное ограниченное поражение в не косметически значимом месте можно лечить либо местно кортикостероидами, ингибиторами кальциневрина, имиквимодом, витамином D, либо с помощью рецидивирующей фототерапии. с ультрафиолетовым (УФ) светом.Все методы имели клинический успех в серии случаев или в неконтролируемых испытаниях, что проявлялось уменьшением гиперпигментации и уменьшением толщины кожи. Многие дерматологи назначают эти препараты в комбинации и рекомендуют использовать окклюзионную повязку для увеличения всасывания местной терапии [86]. УФ-обработка имеет несколько модальностей; широкополосный UVA, узкополосный UVB и UVA-1. UVA-1 был успешным в двух рандомизированных контролируемых исследованиях, особенно в отношении ранних воспалительных и поверхностных кожных поражений [87, 88].Однако доступ к УФ-терапии ограничен, а лечение требует много времени (часто требуется три сеанса по 30–60 минут в неделю), что делает этот вариант сложным для многих. Кроме того, риск потенциальных долгосрочных эффектов, таких как старение кожи и канцерогенез, у детей неизвестен.
Среди ревматологов и дерматологов растет консенсус в отношении того, что системная терапия оправдана при умеренном или тяжелом СЛ. Признаки, которые могут указывать на среднетяжелое или тяжелое заболевание, включают глубокое вовлечение подкожной клетчатки, фасций и мышц, пересекающих сустав и потенциально вызывающих функциональные нарушения, линейные или атрофические поражения, поражающие лицо/кожу головы, быстро прогрессирующее или широко распространенное активное заболевание, а также неэффективна местная или УФ-терапия [86, 89].Наиболее часто используемой системной терапией является метотрексат (МТ) в сочетании с кортикостероидами (КС), что подтверждено недавним опросом 158 педиатрических ревматологов Северной Америки, проведенным Li et al. [89]. Клинические виньетки были розданы клиницистам, описывающим пациентов с умеренно-тяжелым LS, и клиницистов попросили дать свои рекомендации по лечению. Реакция клиницистов на начальную терапию заключалась в подавляющем большинстве случаев в комбинации с метотрексатом и кортикостероидами (более 80% использовали бы оба препарата). Несмотря на то, что было достигнуто отличное согласие в отношении выбора лекарств, схемы лечения широко различались в отношении приема (пероральный или комбинированный).подкожно или внутривенно), а также снижение дозы кортикостероидов и продолжительность терапии [89].
Подкомитет CARRA вырабатывает наилучшие согласованные протоколы лечения для пациентов с умеренным и тяжелым LS, чтобы помочь решить эту проблему и сузить список схем, которые были бы полезны клиницисту. Эти протоколы имеют четыре плана лечения для индукции терапии. Три протокола включают подкожное введение метотрексата (1 мг/кг, максимальная доза 25 мг в неделю), а четвертый был разработан для тех, у кого метотрексат неэффективен или у кого непереносимость метотрексата.Протоколы подразделяются на монотерапию метотрексатом, метотрексат с пероральным КС и постепенной дозировкой, метотрексат с внутривенным КС и микофенолата мофетил (ММФ) в дополнение к пероральному или внутривенному КС. Эти протоколы в настоящее время представляются для публикации. Было показано, что MMF эффективен при тяжелом резистентном к метотрексату педиатрическом LS, о чем свидетельствует серия случаев из 10 пациентов (два с пансклеротическим морфеозом, три с GM, два с LiScl конечностей и 3 с LiScl головы). Во всех случаях наблюдалось улучшение при приеме ММФ и удалось снизить дозу кортикостероидов [90].
Первое двойное слепое рандомизированное контролируемое исследование у детей с LS с использованием объективных показателей было проведено Zulian et al., в котором сравнивались метотрексат и пероральные кортикостероиды с метотрексатом и пероральными кортикостероидами.плацебо и пероральные КС. Было установлено, что метотрексат эффективен и безопасен, что подтверждает поддержку сообществом использования метотрексата при LS. Кроме того, у тех, кто лечился метотрексатом, значительно улучшилось состояние и было значительно меньше рецидивов по сравнению с теми, кто принимал только КС [84].
Другие методы лечения
Интенсивная физиотерапия и трудотерапия в сочетании с системной иммуносупрессивной терапией рекомендуется пациентам с линейной или глубокой склеродермией конечностей, чтобы помочь избежать или свести к минимуму контрактуры суставов.Кроме того, для пациентов с ECDS и / или PRS вмешательство пластической хирургии считается разумным после того, как заболевание находится в стадии ремиссии без каких-либо признаков клинической активности. Аугментация включает в себя растирание жировой ткани микрокаплями из подкожной клетчатки живота пациента, чтобы помочь заполнить часть подкожной атрофии, чтобы получить более симметричный контур лица (). При более тяжелом заболевании кожно-фасциальные лоскуты и реконструкция лицевых костей также являются вариантами, хотя обычно их применяют в позднем подростковом возрасте, после того как скелет созрел [91].Один центр сообщает, что из 10 подростков с ECDS и/или PRS, перенесших хирургическое вмешательство для коррекции лицевой деформации, 9 сообщили об удовлетворении результатами процедуры (процедур), и все они рекомендовали бы хирургическое вмешательство другим людям с этим заболеванием, что означает потенциальную большую пользу. влияние на качество жизни этих пациентов [92].
Синдром Парри-Ромберга, поражающий левое лицо до (А) и после (В) пересадки подкожно-жировой клетчатки.
Детская склеродермия – системная и локализованная формы
Краткое изложение
Детская склеродермия включает две основные группы клинических форм: системный склероз (СС) и локализованную склеродермию (ЛС).Хотя оба имеют общую патофизиологию с начальной воспалительной фазой, связанной с эндотелиальной активацией, и более поздней фиброзной фазой, проявляющейся коллагенизацией ткани и заметной толщиной кожи, их клинические проявления различаются. СЛ, как правило, ограничивается кожей и подкожным слоем, и хотя он не является смертельным, как ССД, до четверти пациентов могут иметь внекожные проявления заболевания, такие как артрит и увеит. Хотя при СС может поражаться любой орган, у детей чаще всего наблюдаются поражения сосудов (феномен Рейно), кожи (утолщение кожи), желудочно-кишечного тракта, легких и опорно-двигательного аппарата.Профили аутоантител при ССД в детском возрасте могут помочь в прогнозировании поражения внутренних органов. Лечение обеих форм склеродермии нацелено на активную воспалительную стадию и останавливает прогрессирование заболевания; тем не менее, все еще необходимо добиться прогресса в разработке более эффективной антифиброзной терапии, чтобы помочь обратить вспять повреждение, вызванное болезнью.
Ключевые слова: системный склероз, локализованная склеродермия, морфеа, детская ревматология
Детская склеродермия
Термин склеродермия буквально означает «склероз», склерозирование или уплотнение «дермы», кожи.«Склеродермия» охватывает обе формы заболевания: системный склероз (СС), характеризующийся фиброзом кожи, сосудов и внутренних органов, который чаще поражает взрослых, и локализованную склеродермию (ЛС), характеризующуюся фиброзом кожи и подлежащих тканей без сосудистых или поражение внутренних органов, которое чаще поражает детей. Хотя они имеют общую основную патофизиологию чрезмерного отложения коллагена при аутоиммунных заболеваниях, эти два состояния клинически различны с уникальными заболеваемостью и прогнозами (2).И то и другое редко встречается у детей: расчетная годовая заболеваемость СЛ составляет 1–3 случая на 100 000 детей [1], а ССД — 1 случай на миллион детей [2]. Средний возраст начала обеих форм детской склеродермии составляет от 7,3 до 8,8 лет. Хотя менее чем у 5% всех пациентов с ССД заболевание начинается в детском возрасте, у большинства пациентов с СЛ оно начинается в детском возрасте [3–6]. К сожалению, существует значительная задержка в диагностике, в среднем от 1,9 до 2,8 лет для ССД [3, 4] и от 1,2 до 1,6 лет для СЛ [5, 7].Для немногих случаев врожденного поражения СЛ среднее время установления диагноза больше, от 3,3 до 3,9 лет [7, 8]. Примерное соотношение женщин и мужчин при ССД у детей составляет 4:1 [3, 4], а при СЛ у детей — 2:1 [5]. Нет четких доказательств расовой предрасположенности к любой из форм детской склеродермии.
Детская склеродермия подразделяется на системную и локализованную болезнь, которая далее дифференцируется на подтипы на основании клинических проявлений поражения кожи. Клинические подмножества связаны с конкретными заболеваниями, как уже упоминалось.
ИЗЛ = интерстициальное заболевание легких; ЛАГ = легочная артериальная гипертензия; ROM = диапазон движений
Системный склероз
Классификация – клиническая и серологическая
Системный склероз (СС) является редким, но потенциально опасным для жизни состоянием. Существует три основных клинических подтипа, связанных с различными заболеваниями (): диффузный кожный (dc) SSc, характеризующийся широко распространенным и быстро прогрессирующим утолщением кожи (распространяющимся проксимальнее локтей и коленей), связанный с ранним поражением внутренних органов (легких, сердца и почек), ограниченный кожный ССД (lcSSc), характеризующийся ограниченным и непрогрессирующим утолщением кожи (ограничивается дистальными отделами конечностей), связанный с поздним заболеванием внутренних органов (легочная артериальная гипертензия, мальабсорбция), и перекрывающийся SSc, который может быть dcSSc или lcSSc с признаками другого заболевания соединительной ткани , такие как дерматомиозит или системная красная волчанка (СКВ) [9].Пациентов с CREST-синдромом (кальциноз, феномен Рейно, нарушение моторики пищевода, склеродактилия и телеангиэктазии) относят к ЛСКС. Некоторые пациенты с недифференцированным или смешанным заболеванием соединительной ткани могут иметь блестящую или полную кожу дистальных отделов пальцев, но у них должен быть основной критерий «уплотнение кожи или склероз проксимальнее пястной кости (MCP) или плюснефаланговой кости (MTP)». считается несовершеннолетним SSc (). Существуют дополнительные два малых критерия, необходимые для диагностики ССД у детей в соответствии с международным консенсусным соглашением () [10].В дополнение к степени утолщения кожи определенные профили аутоантител в сыворотке, связанные со склеродермией, помогают идентифицировать подмножества ССД и прогнозировать их поражение органов как у взрослых, так и у детей с началом ССД [3, 4, 6, 11, 12]. Например, антитело против топоизомеразы (Scl-70) можно ожидать у пациента с dcSSc, и это может вызывать беспокойство в связи с быстрым прогрессированием поражения кожи и развитием интерстициального заболевания легких (ИЗЛ). [3, 4, 6]. Четырьмя наиболее распространенными антителами при ССД у детей являются антитела к топоизомеразе (Scl-70) (20–34%), обычно связанные с dcSSc и ИЗЛ, как уже упоминалось, за которыми следуют антитела к центромерам (ACA) (2–16%). связанный с lcSSc и легочной артериальной гипертензией (ЛАГ), а также антитела к U1-RNP и Pm-Scl (полимиозит-склеродермия) (2–16%), оба из которых обычно обнаруживаются у пациентов с перекрывающимися синдромами SSc и другими соединительными заболевания тканей с более выраженными признаками артрита и миозита, такие как дерматомиозит или системная красная волчанка [3, 4, 6].Положительная доля пациентов с U1-RNP и Pm-Scl, вероятно, выше, чем сообщалось, поскольку некоторые авторы не включают случаи перекрывающегося SSc или смешанных заболеваний соединительной ткани в своих педиатрических когортах SSc. В целом, частота этих антител у детей отражает частоту клинической подгруппы dcSSc, lcSSc и перекрывающегося SSc [3, 4, 6]. По сравнению со взрослым SSc перекрывающийся SSc гораздо чаще встречается у детей (29% против 9%). , что отражается повышенной частотой антител U1-RNP и Pm-Scl и связано с более высоким процентом миозита и артрита, наблюдаемых у детей с ССД [3].Есть несколько других аутоантител, связанных с ССД, которые связаны с различными органными проявлениями; однако они гораздо реже встречаются при СД в детском возрасте. Одним из них является антитело к РНК-полимеразе III (POL3), которое относится к тяжелым почечным заболеваниям в форме склеродермического почечного криза. POL3 редко наблюдается при ССД в детском возрасте, но может быть обнаружен у 30% взрослых пациентов с ССД и отражает клинически значимую более высокую долю почечной недостаточности при ССД у взрослых (3, 6). Несмотря на то, что многие аутоантитела, связанные со склеродермией, были идентифицированы у взрослых ССД, существует высокая доля детей, 20-23%, которые имеют положительный результат АНА без идентифицированных специфических аутоантител [3, 6].
Типичная блестящая и толстая кожа рук с уплотнением кожи, распространяющимся вверх по предплечью (за пястно-фаланговые суставы) у пациента с системной склеродермией.
Таблица 1
Временные рекомендации по классификации Критерии несовершеннолетних системного склероза *
| Основным критерием (обязательно) | Устранение кожи / утолщение проксимальных к MCP или MTP-соединениям | ||
| Незначительный критерий (2 требуется) (Scleroderma конкретный орган участие органов) | |||
| COSCORE | |||
| Sclerodactyly | |||
| Мопиллярные изменения ногтей (мегакапилларии и авашелярные зоны) | |||
| Digital Tip lexers | |||
| Dysphasia | |||
| Gastroesophageal Reffull | |||
| Cardiac | |||
| Arrhhythmias | |||
| Сердце сбой | |||
| почек | |||
| почек | |||
| Scleroderma Renal Crisis | |||
| новая артериальная гипертония | |||
| легочной артериальной гипертензии (первичной или вторичной по отношению к ILD, оценивали по данным эхокардиографии) | |||
| Неврологические | |||
| невропатии | |||
| кистевой туннельный синдром | |||
| Musculoskeletal | |||
| фрикционные трется Сухожильные | |||
| Arthrititis | |||
| серологический | |||
| Антиатомное антитело | |||
| SSC-селективный автоантибоди es | |||
| Антитопоизомераза 1 (Scl-70), антицентромерная, антиРНК-полимераза I или III, анти-PM-Scl, антифибриллин |
Клинические проявления
Основные клинические проявления у детей SSc
Сосудистый
Общими признаками в начале детского SSc являются феномен Рейно (RP) (70%) и кожные изменения рук (60%), включая отек, склеродактилию и уплотнение проксимальнее MCP [4].В наибольшей изученной когорте педиатрических ССД (153 пациента) 53% имели как уплотнение кожи рук, так и РН, а 10% пациентов с РН также имели инфаркты пальцев [4]. На протяжении всего заболевания почти у всех будет РН (97%) и изменения кожи кистей (96%) [3].
Девочка-подросток с типичной (А) склеродактилией и (В) пальцевыми изъязвлениями и язвами.
Феномен Рейно — вазоспастическая реакция, приводящая к трехфазному изменению цвета рук (сначала бледность из-за вазоконстрикции, затем посинение вследствие цианоза и, наконец, краснота из-за реперфузии с последующим отеком и болью), связанная с ощущением онемения и покалывания .RP также может возникать в пальцах ног и других акральных областях, включая уши, кончик носа или язык. При ССД артериолы более узкие и жесткие из-за выраженной гиперплазии интимы и фиброза вследствие эндотелиальной дисфункции, что приводит к более выраженному РП с последующим повреждением/ишемией тканей [13]. Эти васкулопатические изменения идентифицируются в капиллярном ложе ногтевого ложа у этих пациентов с помощью капиллярной микроскопии, демонстрируя дилатацию, извилистость, кровоизлияние, выпадение (аваскуляризацию) и более позднюю разветвленность капилляров [14].Сообщается, что распространенность капиллярных изменений ногтевого ложа при ССД у детей составляет 50% [3, 4], но, вероятно, выше, если проводилась стандартизированная микроскопия. Плохая перфузия пальцев в конечном итоге приводит к изъязвлениям кончиков пальцев, изъязвлению (2), а в более тяжелых случаях — к аутоампутации. Здоровые люди также могут страдать феноменом Рейно без риска развития основного заболевания соединительной ткани (первичный синдром Рейно). У этих людей обычно никогда не развиваются язвы на кончиках пальцев или капиллярные изменения ногтевого валика, поскольку у них нет основной васкулопатии.Признаки, которые помогают клиницисту дифференцировать первичный феномен Рейно от феномена, вторичного по отношению к заболеванию соединительной ткани (ЗСТ), как у детей, так и у взрослых, следующие: отсутствие изменений капилляров ногтевого валика, изъязвление и/или изъязвление кончиков пальцев и отрицательный АНА [15, 16].
;;9 и 2,8 года между первыми признаками заболевания и диагнозом [3, 4]. В начале клинического течения кожа отечна, особенно на дистальных отделах конечностей; редко присутствует поражение проксимальных отделов конечностей, лица и туловища. Фаза индурации, в честь которой названа склеродермия, характеризуется потерей естественной податливости кожи и наличием пальпируемой толщины кожи. Кожа приобретает блестящий, напряженный вид с сужением дистальных отделов пальцев (и ). Со временем, по мере того как кожа утолщается, а нижележащие структуры, такие как сухожилия, поражаются и укорачиваются, суставы пальцев начинают терять диапазон движений как при разгибании, так и при сгибании, а в более тяжелых случаях возникает деформация «когтеобразной руки», что сильно влияет на работоспособность. повседневная деятельность (ADL).Типичное лицо склеродермии с натянутой кожей и атрофией кожи характеризуется сморщенным носом, тонкими сжатыми губами, маленьким ртом, выступающими зубами и невыразительным видом (4). Толщина кожи оценивается как легкая, умеренная и тяжелая по всему телу, и кумулятивный балл получается с использованием модифицированной шкалы кожи Роднана (mRSS) [17]. mRSS получают в ходе лонгитюдных посещений, и можно рассчитать скорость прогрессирования толщины кожи (STPR), что помогает прогнозировать время поражения внутренних органов у пациентов с dcSSc.Высокий STPR связан со склеродермическим почечным кризом [18].
Невыраженное лицо при склеродермии. Девочка препубертатного возраста с уменьшенным ротовым отверстием и «поджатыми губами».
Другие кожные проявления при СС включают подкожные отложения кальция (кальциноз кожи), которые могут возникать в точках давления, обычно обнаруживаемых на разгибательных поверхностях суставов кистей при СС, и могут иногда выдавливаться через кожу подобно дерматомиозиту. Эти поражения могут быть болезненными и изъязвляться.Однако они чаще связаны с хроническим заболеванием и редко присутствуют во время начала заболевания и постановки диагноза. Телеангиэктазии также являются проявлением ССД, чаще всего встречаются в варианте lcSSc (ранее известном как CREST-синдром) и обычно локализуются на лице и верхних конечностях.
Другие органные проявления при ССД (в порядке убывания частоты) включают желудочно-кишечные, легочные, скелетно-мышечные, сердечные, почечные и неврологические симптомы. Желудочно-кишечные симптомы встречаются примерно у половины детей, хотя более детальное исследование часто указывает на наличие аномалий у большего процента [3].Нарушение моторики пищевода и гастроэзофагеальный рефлюкс часто приводят к дисфагии и эзофагиту. Дистальная дисфагия, особенно при приеме твердой пищи, вызывающая ощущение «застревания пищи» в середине грудной клетки, является типичной и представляет собой дисфункцию дистальной гладкой мускулатуры пищевода. Гастропарез с замедленным опорожнением желудка в дополнение к дисфункции пищевода может привести к усилению симптомов рефлюкса. Обычно пациенты жалуются на ночной кашель в положении лежа на животе из-за тихой аспирации и солоноватый привкус во рту при пробуждении из-за тихого рефлюкса.Хронический рефлюкс может привести к стриктурам пищевода. Оценка желудочно-кишечного тракта с использованием манометрии и внутрипищеводного 24-часового рН-мониторинга считается чувствительным индикатором сниженного тонуса пищевода и рефлюкса [19]. Однако в педиатрии эти методы используются реже. Вместо этого обычно используется исследование глотания для оценки аспирации и нарушений моторики с последующим исследованием верхних отделов желудочно-кишечного тракта с последующим исследованием тонкой кишки. Если поражена тонкая кишка, спазмы, диарея и запор могут быть результатом перистальтической дисфункции.Эпизоды псевдообструкции с постпрандиальным вздутием живота, болью и тошнотой могут возникать из-за функциональной кишечной непроходимости. Могут наблюдаться избыточный бактериальный рост, стеаторея, потеря веса, заворот и даже перфорация. Заболевание толстой кишки проявляется в виде широкоротых дивертикулов и потери нормальной архитектуры толстой кишки. Другим осложнением поражения ЖКТ при ССД является острое желудочно-кишечное кровотечение, связанное с эктазией антральных вен желудка (ГАВЭ), требующее фотокоагуляции. Это состояние редко встречается у детей и связано с ранним dc SSc, РНК-полимераза III положительных пациентов [20].
Поражение легких при педиатрическом ССД составляет в литературе от 30 до 70% и включает интерстициальное заболевание легких (ИЗЛ), легочную артериальную гипертензию (ЛАГ) (либо первичную, которая возникает в результате васкулопатии, либо вторичную из-за ИЗЛ), и аномальные легочные функциональные тесты (PFTs). Особое беспокойство вызывают аномалии PFT, такие как снижение форсированной жизненной емкости легких (ФЖЕЛ) и диффузионной способности монооксида углерода (DLCO) [21]. Интерстициальное заболевание легких (ИЗЛ) обычно начинается с воспалительной фазы, авеолита, характеризующегося смешанным клеточным инфильтратом в интерстиции легкого, который распространяется в альвеолярные пространства.Бронхоавеолярный лаваж (БАЛ) демонстрирует увеличение нейтрофилов и/или эозинофилов в дополнение к альвеолярным макрофагам [22]. На наличие авеолита также указывают затемнения по типу матового стекла на компьютерной томографии высокого разрешения (КТВР) легких. По мере прогрессирования ИЗЛ воспаление стихает, так как происходит переход в сторону фиброза с утолщением альвеолярных стенок и ремоделированием легкого. Прогрессирующий легочный фиброз может привести к рестриктивному заболеванию легких от умеренной до тяжелой степени со значительным снижением ФЖЕЛ и DLCO.Клинически ИЗЛ обычно проявляется в виде медленно прогрессирующей одышки при физической нагрузке в течение многих лет. Для сравнения, ЛАГ проявляется быстрым прогрессированием одышки при физической нагрузке в течение нескольких месяцев. В условиях ЛАГ PFT демонстрируют снижение DLCO и аномальные результаты эхокардиограммы с предполагаемым систолическим давлением в легочной артерии (ЛА) или правом желудочке (ПЖ)> 40 мм рт.ст., что подтверждается катетеризацией правых отделов сердца со средним давлением ЛА> 25 мм рт.ст. Для большинства детей с ССД или подозрением на ССД первоначальная оценка включает рентгенографию грудной клетки, тесты функции легких, включая DLCO, и эхокардиограмму.Дополнительные сердечно-легочные тесты проводятся, если обнаруживаются аномалии или пациент слишком молод, чтобы надлежащим образом завершить тестирование (например, PFT). Аномальные PFT требуют оценки с помощью HRCT грудной клетки и, возможно, BAL, если требуются дополнительные доказательства воспаления (авеолит) или необходима оценка инфекции перед началом терапии [21, 23]. Признаки ЛАГ на эхокардиограмме более формально оцениваются при катетеризации правых отделов сердца.
По сравнению с началом ССД у взрослых поражение опорно-двигательного аппарата чаще встречается у детей с ССД.Приблизительно 30–40% страдают воспалительным артритом (суставные выпоты) в дополнение к типичному «сухому синовиту» склеродермии, выражающемуся в фиброзе сухожилий, пересекающих суставы, что ограничивает диапазон их движений (ROM). В начале патологического процесса, особенно у пациентов с dcSSc, теносиновит/бурсит вызывает ощутимый шум трения сухожилий («кожистый хруст») при разгибании или сгибании сустава [24]. При миопатии у больного обычно отмечается симметричная проксимальная слабость, особенно плечевого пояса и плечевых мышц, иногда с выраженной атрофией.Миозит также проявляется повышением уровня мышечных ферментов и изменениями групп мышц на МРТ, сходными с таковыми при ювенильном дерматомиозите (ЮДМ). Биопсия мышц при ССД отличается от таковой при ЮДМ наличием большего количества фиброза и наличием утолщенных капилляров [25]. Дети с dcSSc и миозитом особенно подвержены риску тяжелого поражения сердца, включая дефицит перфузии миокарда и дилатационную кардиомиопатию [26].
Поражение сердца , хотя и редко, но является основной причиной связанной со склеродермией смертности у детей с ССД, о чем свидетельствуют 10 из 15 смертей в когорте Martini et al. [4] и 5 из 32 смертей в Scalapino et al. когорта [3].Сердечные проявления являются результатом сочетания миокардиального фиброза, сосудистой недостаточности и воспаления и включают сердечную блокаду, аритмию, застойную сердечную недостаточность, кардиомиопатию и перикардит. В отличие от детского ССД, смертность от склеродермии у взрослых в первую очередь обусловлена поражением легких, как ИЗЛ, так и ЛАГ [27].
Легкая почечная дисфункция не редкость при ССД и является вторичной по отношению к васкулопатии, а не к гломерулонефриту, который наблюдается при системной красной волчанке.Это подтверждается патологическими данными пролиферации интимы междольковых артерий почек и отсутствием активного мочевого осадка (т.е. эритроцитарных цилиндров). Напротив, более тяжелая и болезненная форма заболевания почек, склеродермия , почечный криз (SRC), встречается только примерно у 15% взрослых пациентов с ССД и еще реже у детей. СРК характеризуется ускоренной артериальной гипертензией, почечной недостаточностью, микроангиопатической гемолитической анемией и тромбоцитопенией.При SSc у взрослых SRC ранее имел 20% выживаемость в течение 1 года, но теперь выживаемость составляет 80% из-за появления ингибиторов ангиотензинпревращающего фермента (ACEI) [27]. SRC чаще всего наблюдается на ранних стадиях заболевания у пациентов с РНК-полимеразой III dcSSc и, как полагают, усиливается при более высоких дозах кортикостероидов. Почечный криз у детей с ССД встречается редко, с менее чем 5% случаев, и только 5 случаев зарегистрированы (2 из которых умерли от почечной недостаточности) среди трех больших опубликованных когорт [3, 4, 28].
Исход
Несмотря на все возможные поражения органов при ССД у детей, она имеет гораздо более благоприятный прогноз через 5, 10 и 15 лет по сравнению со ССД у взрослых с более низкой частотой тяжелого поражения органов. Foeldevari и коллеги [28] недавно сообщили, что показатели выживаемости Kaplan Meier в течение 10 лет для детей составляют 98% по сравнению с 75% у взрослых. Лечение ССД зависит от поражения органов, но общие меры, такие как антациды для защиты ЖКТ, прокинетики для нарушения моторики, чередование антибиотиков для мальабсорбции, а также сосудорасширяющие средства при феномене Рейно (RP) (могут также помочь при ЛАГ), нестероидные Обычно рекомендуются противовоспалительные препараты (НПВП) и физиотерапия (ЛТ) при артрите или тендините.Кортикостероиды при миозите, артрите и других воспалительных клинических проявлениях следует использовать с осторожностью, поскольку они могут спровоцировать склеродермический почечный криз. Более конкретные рекомендации по лечению различных органных проявлений были согласованы на консенсусной конференции международных экспертов по склеродермии и опубликованы в 2009 г. [29]. Примечательно, что в лечении раннего диффузного кожного заболевания как у детей, так и у взрослых произошел переход от использования D-пеницилламина к микофенолата мофетилу (ММФ) из-за потенциальных антипролиферативных эффектов, хотя это не было включено в рекомендации по лечению, которые обсуждались ранее. 30].
Локализованная склеродермия
Классификация
Локализованная склеродермия, также известная как «морфея», имеет другой тип поражения кожи, чем системный склероз (СС), и включает несколько подтипов, классифицированных по глубине и характеру поражения(й). Группа заболеваний характеризуется фиброзом, который в основном ограничивается кожей и подкожной клетчаткой; однако более глубокие формы также включают фасции, мышцы, сухожилия и суставную капсулу. Традиционная классификация, предложенная Peterson et al.[31] описали пять подклассов, включая бляшечную морфею, генерализованную морфею, буллезную морфею, линейную морфею, глубокую и пансклеротическую морфею (1). Однако совсем недавно консенсус экспертов внес изменения, чтобы предоставить более клинически применимые классификации, названные «Критерии Падуи», которые включают подтип «смешанного морфеа», учитывая, что распространенность этой категории составляет 15% () [32].
Таблица 2
| A: LS ° Подтипы: Mayo Классификация ‡ |
|---|
| Налет Morphea † |
| Morphea ан бляшек † |
| каплевидный Morphea |
| атрофодермия из Pasini и Пьерини |
| Обобщенная Morphea † |
| Буллезная Morphea |
| линейный Morphea † |
| линейная Morphea (линейная склеродермия) † |
| En купе де сабля † |
| Progressive гемифациальный атрофия |
| Deep Morphea † |
| Подкожный Morphea † |
| эозинофильный фасциит | Morphea profunda † |
| Отключения pansclerotic Morphea детей |
| б: Предлагаемые подтипов для ювенильного LS °: Падуя Предварительной Классификации ‡ | |||
|---|---|---|---|
| Описанного Morphea | Овальные/круглые поражения
| 1||
| Linear Scleroderma | Линейные поражения могут включать дерма, подкожная клетчатка, мышцы, кости
| Обобщенные Morphea | ≥ 4 Большие бляшки (> 3см) по крайней мере 2 7 анатомических областей: голова/шея, правая верхняя конечность, LUE, RLE, LLE, передняя часть туловища, задняя часть туловища | |
| Пансклеротическая морфеа | Окружное поражение конечности (за исключением кончиков пальцев рук и ног) Все глубины кожи/подкожной клетчатки /мышца/кость | ||
| Смешанная форма морфеа | Комбинация двух или более вышеперечисленных подтипов | ||
Активность заболевания и признаки повреждения (клинические и гистологические данные, клинические и гистологические признаки) Сходны с СЛ
более ранняя более активная фаза заболевания и более поздняя более фиброзная фаза ().Ранние активные поражения при СЛ характеризуются фиолетовой воспалительной каймой или «сиреневым кольцом» (1) и индурацией кожи по всему очагу поражения, включая границу. Во время этого активного состояния поражения также могут увеличиваться, и могут накапливаться новые поражения [33]. Гистологическая картина на ранних стадиях заболевания состоит из периваскулярного инфильтрата преимущественно из лимфоцитов с примесью редких плазматических клеток и эозинофилов в глубоком ретикулярном слое дермы и подкожной клетчатке, сопровождающемся утолщением коллагеновых пучков, уменьшением эластических волокон и набуханием эндотелиальных клеток [34, 35].С течением времени поражение болезнью накапливается и проявляется увеличением толщины кожи, особенно в центре поражения, иногда оставляя склеротический очаг цвета слоновой кости (). Гистологическая оценка кожи выявляет утолщенный гипоцеллюлярный (гомогенизированный) коллаген, который заменил прежний воспалительный инфильтрат вокруг придатков дермы, оставив после себя атрофические эккринные железы и волосяные фолликулы. Кроме того, уменьшается количество капилляров с фиброзной стенкой и суженным просветом [34, 35].Клинический результат этих находок отражается в кожной и подкожной атрофии. При физикальном осмотре, когда дерма атрофична, наблюдается отсутствие роста волос в месте поражения, видимые вены и легкий вид «обрыва» на коже (). Подкожная атрофия существует в непрерывном диапазоне и может быть как легкой, как уплощение кожи, так и более «выемчатым» вогнутым видом жировой ткани при более умеренных поражениях, а в самых тяжелых случаях до такой степени, что можно увидеть мышцы, движущиеся под кожей.Поствоспалительная гипер- и гипопигментация также являются результатом предшествующего воспаления и часто являются признаками, которые привлекают внимание врача к поражению. Часто семьи пропускают раннюю активную фазу и считают эритему/фиолетовый цвет скорее синяком или травмой, но именно тот факт, что «синяк, который не проходит» (поствоспалительная гиперпигментация), часто побуждает семью искать медицинская помощь ().
Фазы локализованной склеродермии и связанные с ней признаки.Признаки активного заболевания, эритема, уплотнение/отек кожи и новые или увеличивающиеся очаги поражения возникают первыми и уступают место признакам поражения заболеванием, диспигментации, атрофии и утолщению кожи.
Типичное «сиреневое кольцо», наблюдаемое на границе множественных бляшек на туловище, указывающее на активное заболевание, любезно предоставлено Еленой Поуп из Children’s Sick Kids, Торонто.
Восковидно-желтый очаг поражения в центре бляшки, остающийся на правой стороне шеи спустя годы после начала локализованной склеродермии.
Кожная атрофия LS, проявляющаяся в виде «капельных» поражений у девочки-подростка с генерализованной морфеей.
Патогенез (общий для ССД и ССД)
Общие гистологические данные на коже пациентов с ССД и СЛ иллюстрируют их общую патофизиологию, несмотря на различные клинические особенности заболевания. Оба являются аутоиммунными заболеваниями с воспалительным, фиброзным и сосудистым компонентом. Популяции мононуклеарных клеток, идентифицированные как в образцах кожи SSc, так и в LS, представляют собой преимущественно Т-клетки, как CD8, так и CD4-положительные, с более высоким процентом клеток CD4 (T-хелперы) [36, 37]. Ассоциированные с Th-клетками цитокины и хемокины, такие как интерлейкины [IL]-2, -4, -6, -8, -13 и TNF-a, были идентифицированы в циркулирующей сыворотке и стимулированных PBMC у пациентов с LS и SSc [38–38]. 43].Следует отметить, что некоторые сывороточные цитокины были обнаружены в более высоких концентрациях у пациентов с LS по сравнению с СС, что интересно, поскольку LS называют «локализованным заболеванием»; однако это может подтверждать высокую частоту внекожных проявлений (примерно 25%), продемонстрированную при СЛ [44]. Было выявлено несколько корреляций между клиническими переменными тяжести заболевания LS и цитокинами и их растворимыми рецепторами, такими как количество бляшек и линейных поражений (IL-13, TNF-a, sIL-2r, sIL-6r), генерализованная морфия и субпопуляции линейной склеродермии (IL-2,-4,-6,-8,-13, TNF-a, sIL-2r, sIL-6r) и корреляция антител, антигистоновые и/или оц-ДНК антитела (IL -4, -6, TNF-a, sIL-2r, sIL-6r) [38–40, 45–47].Эти ассоциированные с Т-клетками цитокины, в свою очередь, дополнительно стимулируют фибробласты и эндотелиальные клетки к выработке трансформирующего фактора роста бета (TGF-β) и фактора роста соединительной ткани (CTGF), которые считаются основными стимуляторами тканевого фиброза (за счет увеличения коллагена). продукция) и повреждение эндотелиальных клеток (через влияние молекул адгезии ICAM-1 и VCAM-1) [48–51].
Микрохимеризм является еще одной интересной областью, изучаемой в отношении общего патогенеза LS и SSc.Клинически и гистологически LS и SSc имеют общие характеристики с хронической болезнью «трансплантат против хозяина» (cGVHD) (1), типом микрохимеризма, который играет роль в патогенезе. Химерные клетки были идентифицированы у пациентов с LS и SSc с использованием методов ПЦР в реальном времени [52, 53].
Линейное поражение туловища (A) и ног (B) у ребенка раннего возраста со склеродерматозной хронической болезнью «трансплантат против хозяина».
Аутоиммунитет, как один из компонентов распространения LS, подтверждается профилем аутоантител, сопутствующими сопутствующими аутоиммунными заболеваниями и семейным анамнезом аутоиммунных заболеваний, наблюдаемых у пациентов с LS.Аутоантитела обычно положительны при LS, включая антиядерные антитела (ANA), антигистоновые антитела (AHA) и антитела к одноцепочечной ДНК (ss-DNA Ab) [54]. ANA является классическим серологическим маркером аутоиммунного заболевания и обнаруживается у пациентов с СЛ с высокой частотой, в диапазоне от 42 до 73% [5, 55]. Считается, что более высокие титры связаны с ранним развитием заболевания [55] и повышенным риском внекожных проявлений [44, 56]. Распространенность оц-ДНК при СЛ составляет примерно 50% [57] и связана с линейной склеродермией (особенно с контрактурами суставов), вовлечением глубоких мышц и увеличением количества анатомических участков; все это отражает увеличение тяжести заболевания [56, 58].Было показано, что в подгруппе пациентов с LS одноцепочечное антитело к ДНК отражает статус активности заболевания, при этом титры снижаются по мере клинического улучшения кожных поражений [54, 56]. АГА встречается с аналогичной частотой при СЛ (47%) [59] и также коррелирует с показателями тяжести заболевания; включая общую площадь вовлеченной поверхности тела, количество поражений, линейное заболевание и наличие контрактур суставов [54, 56]. В исследовании, посвященном линейной склеродермии, сочетание положительных ss-ДНК и AHA было обнаружено у 23% пациентов (взрослых и детей с СЛ) и достоверно коррелировало с наличием контрактур суставов [56].Вышеупомянутые антитела указывают на тяжесть заболевания и могут помочь в стратификации пациентов с ранним диагностированным заболеванием, предполагая возможное начало более агрессивной терапии и усиленное наблюдение.
Сопутствующие и семейные ревматические и аутоиммунные заболевания более распространены при СЛ (детском и взрослом) по сравнению с общей популяцией [60]. Сопутствующее заболевание было зарегистрировано у 5–10% детей [7, 44, 60] и 30% взрослых с СЛ [60]. Наиболее распространенными заболеваниями были дерматологические аутоиммунные заболевания (псориаз, витилиго, очаговая алопеция), за которыми следовали ревматические состояния (ювенильный идиопатический артрит, ревматоидный артрит (РА), системная красная волчанка (СКВ) и синдром Шегрена) [7, 60].При оценке по подтипу СЛ частота сопутствующего аутоиммунного заболевания была значительно выше у пациентов с генерализованным морфеозом [60]. Семейные ревматические заболевания и другие аутоиммунные заболевания были зарегистрированы у 12-25% всех пациентов (как у детей, так и у взрослых), наиболее частым из которых был псориаз, за которым следовали РА, СКВ, ССД, СЛ, рассеянный склероз и тиреоидит [5, 7]. , 60]. Вновь была продемонстрирована связь между генерализованной морфеей и повышенной частотой этих заболеваний [5].
Клинические проявления
Хотя термин «локализованная склеродермия» или «морфея» объединяет все подтипы в одну категорию, их кожные/подкожные проявления и внекожные ассоциации совершенно уникальны. Линейная склеродермия (LiScl) является наиболее распространенным подтипом, встречающимся у 50–60% детей с LS, и характеризуется линейной полосой или полосой, поражающей дерму, подкожную клетчатку и часто подлежащие мышцы, сухожилия и кости. LiScl обычно возникает в виде одиночной линейной односторонней полосы на конечностях, туловище или голове (5, 7).Наиболее часто поражаются ноги, за ними следуют руки, лобная часть головы и туловище. Поражения LiScl, поражающие конечности, вызывают наибольшее беспокойство, когда они пересекают сустав, поскольку существует вероятность склероза и уплотнения нижележащих мышц, сухожилий и суставной капсулы, что приводит к контрактуре сустава и связанной с этим дисфункции конечностей. Особое беспокойство у детей с LiScl вызывает поражение эпифизарной зоны роста, что может привести к необратимому укорочению или атрофии конечности.Ортопедические осложнения регистрируются примерно у 30–50% пациентов с LiScl конечностей (конечностей) [7, 58, 61]. Peterson и соавторы сообщили о легкой или умеренной инвалидности при последующем наблюдении у пациентов с LiScl конечностей в их когорте (среднее время наблюдения 9,2 года) [1].
Линейная склеродермия, затрагивающая одностороннее поражение правой верхней конечности (А). У этой молодой женщины ассоциированные контрактуры лучезапястных, пястных и фаланговых суставов. Суставные контрактуры кисти отчетливо видны по сравнению с контралатеральной стороной при активном разгибании (В) и сгибании (С).
Приблизительно у 25% пациентов с LiScl наблюдается поражение волосистой части головы [5]. Линейные поражения кожи головы и лба называются «удар саблей» (ECDS), потому что линейная депрессия напоминает шрам от саблевидной раны. Прогрессирующая гемифациальная атрофия, также известная как синдром Парри-Ромберга (PRS), представляет собой еще один линейный вариант LS, вызывающий гемиатрофию лица, включая нижнюю, верхнюю челюсть, язык, подкожную и мышечную ткани (4). Как правило, ECDS ассоциируется с кожными проявлениями диспигментации и толщины кожи, тогда как PRS не является и обычно имеет более выраженную подкожную атрофию.Эти расстройства могут возникать вместе, либо в одном и том же месте (сторона лица), либо на противоположных сторонах [62]. Несмотря на противоречивость, ECDS и PRS, вероятно, представляют собой разные спектры одного и того же состояния, что подтверждается сходными проявлениями и преобладанием неврологической симптоматики [62, 63] и клеточных инфильтратов (лимфоцитарный инфильтрат сосудисто-нервных пучков) кожи [64] и ткани головного мозга [65]. ]. PRS и ECDS несут одинаковый риск неврологического поражения, при этом у 8–20% пациентов с этими состояниями наблюдается поражение центральной нервной системы, выражающееся в судорогах, хронических головных болях, неврите зрительного нерва и, реже, в нервно-психических изменениях или ишемическом инсульте [62, 63]. ].Также сообщалось об ортодонтических и офтальмологических аномалиях с одинаковой частотой при обоих состояниях [63]. Если начало заболевания (ECDS или PRS) происходит в возрасте до 10 лет, существует особый риск скелетной гипоплазии пораженной стороны лица из-за незрелости скелета, что приводит к стоматологическим и косметическим аномалиям [66]. ].
Саблезубое поражение (ECDS), поражающее волосистую часть черепа и голову односторонним образом (A), вызывающее умеренную потерю подкожной ткани и деформацию скелета (B).
Тяжелый пример синдрома Парри-Ромберга/гемифациальная атрофия правого лица.
Поверхностная ограниченная морфия, ранее называемая бляшечной морфеей по классификации Петерсона, является наиболее частым проявлением СЛ у взрослых (60%) и составляет примерно одну четверть СЛ у детей [5, 60]. Он характеризуется одной или несколькими овальными или округлыми участками уплотнения с восковым центром цвета слоновой кости и фиолетовым ореолом по периферии диаметром от 1 до 30 см. Эти поражения обычно возникают на туловище или проксимальных отделах конечностей (4).Поверхностный вариант ограничен эпидермисом и дермой. Глубокая ограниченная морфеа, ранее называемая подкожной морфеей, глубокой морфеей или глубокой морфеей, представляет собой круглое/овальное поражение, расположенное глубже в ткани. Эти поражения поражают дерму и подкожную клетчатку с различным проникновением в фасции и мышцы. Как правило, имеются только тонкие кожные изменения (такие как небольшая эритема и истончение дермы), но присутствует большое количество воспалительных инфильтратов в глубоких подкожных тканях и фасциях, вызывающих депрессию в жировой ткани («выемчатый» вид) и утолщение. , стянутая кожа.
Типичное изолированное овальное поражение на туловище, соответствующее поверхностным очерченным морфеям.
Генерализованная морфеа (ГМ) определяется наличием более 3 бляшек, каждое из которых имеет ширину более 3 см. Обычно бляшки сливаются и поражают несколько анатомических областей, чаще всего на туловище. ГМ — редкий подтип, встречающийся менее чем у 10% пациентов с СЛ [5, 7]. Как упоминалось ранее, этот подтип часто связан с несколькими аутоиммунными явлениями, включая антитела к одноцепочечной ДНК и AHA, а также сопутствующий и семейный анамнез аутоиммунных заболеваний.Системные симптомы, такие как миалгия, артралгия и утомляемость, чаще встречаются при ГМ [7, 60].
Пансклеротическая морфеа — редкая разновидность СЛ, поражающая только 1–2% пациентов и обычно приводящая к инвалидности. Первоначально он поражает конечности, но позже может мигрировать на туловище, не затрагивая только лицо и дистальные области пальцев рук и ног (4). Отмечается быстро прогрессирующий фиброз глубоких слоев дермы, подкожной клетчатки, фасций и мышц, иногда вовлекающий кости. Это приводит к значительной контрактуре суставов, атрофии мышц, изъязвлениям кожи и иногда к рестриктивной дыхательной недостаточности, если задействован грузовик.Развитие плоскоклеточного рака является дополнительным осложнением при пансклеротической морфее, особенно в области хронических ран [67].
Пансклеротическая морфеа, поражающая конечности по окружности и не затрагивающая дистальные отделы пальцев ног и пальцев.
Смешанная форма морфеа является последним подтипом, рассмотренным на основе консенсуса Падуи, и определяется наличием более чем одного клинического подтипа у одного пациента с СЛ. Смешанная форма морфеа довольно распространена и наблюдается примерно у 15% детей с СЛ.Наиболее частым проявлением смешанного морфеа является сочетание линейного и бляшечного морфеа [5].
Буллезный морфеоз иногда считают редким подтипом СЛ. Тем не менее, многие эксперты согласны с тем, что буллезные поражения являются вторичными по отношению к агрессивному линейному или глубокому СЛ и, вероятно, отражают нарушение лимфатического оттока. Эозинофильный фасциит (ЭФ), когда-то считавшийся типом глубокого морфеа по критериям Петерсона, не считается подтипом СЛ по критериям Падуи. Он часто ведет себя как смесь глубокой морфеи и линейной склеродермии, что приводит к глубокой инфильтрации и контрактурам суставов [31].)
Внекожные проявления
Внекожные проявления (ВКМ) нередки при СЛ и возникают примерно у четверти пациентов на протяжении всего заболевания [44]. Согласно большому международному исследованию 750 детей с LS, внеклеточное маточное окошко чаще встречается у пациентов с LiScl и в основном состоит из ортопедических осложнений (19% в целом в группе, 50% у детей с LiScl конечностей), неврологических или глазных нарушений (5% всей группе, 40% у лиц с LiScl головы), феномен Рейно (2.1% всей группы) и другие аутоиммунные состояния, такие как тиреоидит (2% всех подтипов) [44]. Менее распространенными ЭКМ являются желудочно-кишечные, респираторные и почечные, все они встречаются менее чем у 2% из 750 обследованных пациентов [44]. Эти оценки могут быть ниже, чем истинный процент ECM, поскольку в анализ были включены только пациенты с симптомами. Другим ограничением исследования было отсутствие контрольной популяции для оценки распространенности этих проявлений в сопоставимой детской популяции.В другом исследовании, в котором изучались желудочно-кишечные проявления у пациентов с СЛ (такие как гастроэпофагеальный рефлюкс) и изменения неврологической визуализации у пациентов с ECDS/PRS, исследователи обнаружили более высокую частоту внеклеточного матрикса; 20% и 60% соответственно [63, 68]. Интересно, что в исследовании Zulian et al., включавшем 750 детей с LS, из 168 пациентов с внеклеточным матриксом у 25% были суставные, неврологические и глазные проявления, не связанные с местом поражения кожи, т.е. артрит периферического сустава с непораженной вышележащей кожей и подкожной клетчаткой. , что еще раз подтверждает гипотезу о том, что «локализованная склеродермия» является более системным процессом [44].У 24 из 750 детей (3,2%) было выраженное поражение глаз, у большинства из которых была линейная склеродермия головы (67%) [69]. Эти результаты подтверждают рутинный офтальмологический скрининг пациентов с LS на наличие признаков воспаления глаз, особенно у пациентов с линейной склеродермией головы. Этот безопасный инструмент скрининга может выявить клинически «тихую» болезнь, а лечение может предотвратить долгосрочные последствия для зрения.
Оценка
Ни один лабораторный тест не является диагностическим для LS. Несмотря на «локализованное» заболевание, было выявлено несколько аутоантител, связанных с LS, которые могут помочь в диагностике и указать на более тяжелое/вовлеченное заболевание (, см. раздел об аутоиммунитете ).В частности, положительность ANA, AHA и одноцепочечной ДНК обнаруживалась с относительно высокой частотой (40–50%). Кроме того, ревматоидный фактор присутствует у трети больных, особенно у больных ГМ, а также коррелирует с тяжестью заболевания (количеством поражений) [5]. Общие маркеры воспаления, включая скорость оседания эритроцитов и уровень сывороточного иммуноглобулина, могут быть полезными маркерами активности заболевания у отдельных пациентов с СЛ, особенно с глубокими или эозинофильно-подобными вариантами.Однако у большинства пациентов с активными поражениями LS эти маркеры не повышены. Диагноз LS обычно ставится при клиническом обследовании, и, если есть сомнения, биопсия кожи и подкожной клетчатки может помочь в диагностике. Дифференциальная диагностика ограничена и включает два состояния кожи с хорошо выраженными бляшечными поражениями: склерозирующий и атрофический лихен и атрофодермию Пазини и Пьерини. Оба являются поверхностными дермальными и эпидермальными воспалительными состояниями, которые, по мнению некоторых, относятся к более мягкому спектру локализованной склеродермии.В дифференциальный диагноз включают кожную Т-клеточную лимфому, особенно с более глубоким багровым изменением цвета кожи, и коллагеному, обычно проявляющуюся уплотнением кожи и подкожного слоя без пигментных изменений с рождения или в раннем детстве. Другие возможные состояния связаны с определенным воздействием и, как правило, имеют более широко распространенные поражения, такие как реакция «трансплантат против хозяина» (состояние после трансплантации), нефрогенная фиброзирующая дерматопатия (гадолиний вводят при нарушении функции почек) и поздняя кожная порфирия (воздействие солнца).
Для оценки поражений кожи и подкожной клетчатки при LS используется несколько инструментов, таких как клиническая оценка кожи, инструменты измерения и визуализация.Клинические оценки кожи обычно используются врачами и включают модифицированную шкалу Роднана [17] и инструмент оценки локализованной склеродермии кожи (LoSCAT), который объединяет параметры активности заболевания и повреждения [33, 70]. Компьютеризированная оценка кожи (CSS) может использоваться для объективного определения размера поражения [71]. Инструменты измерения, в том числе дюрометр, который измеряет степень твердости кожи [72], и кутометр, который измеряет эластичность кожи [73], также были предложены в качестве полезных.Кроме того, для диагностики или оценки можно использовать несколько типов визуализации, таких как термография [74, 75], лазерная допплеровская флоуметрия [76], УЗИ [77, 78] и МРТ [79]. Некоторые из этих методов были проверены в LS, включая LoSCAT, CSS, ультразвук с использованием более высоких частот (10–25 МГц) и МРТ. Однако в настоящее время стандартные меры широко не используются, и оценка зависит от методов, специфичных для центра, которые зависят от опыта исследователя.
Недавние усилия Альянса по исследованиям детского артрита и ревматологии (CARRA) были сосредоточены на совместных проектах исследователей детской ревматологии и дерматологии в Северной Америке.Их цель состоит в том, чтобы установить конкретные клинические меры для оценки активности и повреждения, которые помогут в мониторинге прогрессирования и состояния болезни при LS. Эти проекты находятся в процессе оценки клинических показателей, таких как LoSCAT, и определения других с помощью многоцентровых исследований с группой клинических и ультразвуковых исследований локализованной склеродермии (LOCUS). Группа LOCUS состоит из экспертов по СЛ, как детских ревматологов, так и дерматологов, которым было предложено ранжировать клинические переменные, наиболее сильно отражающие активность заболевания и повреждения при СЛ.Консенсусное согласие >75% и высокая содержательная достоверность (модифицированная каппа-статистика 0,88–1,00) в отношении четырех параметров активности; появление новых поражений, расширение существующих поражений, эритематозный/фиолетовый оттенок на границе и уплотнение кожи на границе поражения [33]. Параметры повреждения кожных заболеваний, получившие наивысшее место среди группы, включали поствоспалительную гиперпигментацию и гипопигментацию, кожную и подкожную атрофию (согласие 88% и межпунктовый индекс достоверности 0.88 – 1,0) [70].
После начала системной иммуносупрессивной терапии у пациентов с умеренным и тяжелым СЛ признаки активности заболевания обычно исчезают в течение нескольких месяцев, а некоторые признаки повреждения уменьшаются от легкой до умеренной степени [80–85]. Именно после этого начального улучшения важно выявление субклинического заболевания, чтобы не отменять иммуносупрессию слишком рано, что может привести к рецидиву [80, 83, 84]. Недавнее исследование педиатрического СЛ выявило 28% рецидивов после средней продолжительности лечения метотрексатом, равной 2.4 года. Было обнаружено, что линейный подтип конечностей и пожилой возраст в начале заболевания являются значимыми предикторами рецидива, и, хотя это не значимо, была заметная разница в продолжительности лечения метотрексатом между рецидивирующими и нерецидивирующими пациентами [85]. Тем не менее, по-прежнему трудно обнаружить субклиническое заболевание, поэтому используются различные методы визуализации и тепловизионного исследования.
Лечение
Подход к лечению СЛ изначально зависит от тяжести заболевания, которое характеризуется подтипом СЛ, возможным вовлечением более глубоких тканей и структур (таких как суставы), локализацией СЛ и степенью активности заболевания.Продолжительность заболевания действительно влияет на состояние активности болезни, при этом большинство поверхностных ограниченных или бляшечных поражений «выгорают» через три-пять лет. Любой оставшийся центральный склероз может со временем смягчиться без вмешательства. Если остаются только параметры поражения болезнью и нет расширения, эритемы или других клинических параметров активности заболевания, допустимо простое наблюдение за поражением. Однако, если есть какие-либо признаки активного заболевания, как детские ревматологи, так и дерматологи согласны с тем, что одно поверхностное ограниченное поражение в не косметически значимом месте можно лечить либо местно кортикостероидами, ингибиторами кальциневрина, имиквимодом, витамином D, либо с помощью рецидивирующей фототерапии. с ультрафиолетовым (УФ) светом.Все методы имели клинический успех в серии случаев или в неконтролируемых испытаниях, что проявлялось уменьшением гиперпигментации и уменьшением толщины кожи. Многие дерматологи назначают эти препараты в комбинации и рекомендуют использовать окклюзионную повязку для увеличения всасывания местной терапии [86]. УФ-обработка имеет несколько модальностей; широкополосный UVA, узкополосный UVB и UVA-1. UVA-1 был успешным в двух рандомизированных контролируемых исследованиях, особенно в отношении ранних воспалительных и поверхностных кожных поражений [87, 88].Однако доступ к УФ-терапии ограничен, а лечение требует много времени (часто требуется три сеанса по 30–60 минут в неделю), что делает этот вариант сложным для многих. Кроме того, риск потенциальных долгосрочных эффектов, таких как старение кожи и канцерогенез, у детей неизвестен.
Среди ревматологов и дерматологов растет консенсус в отношении того, что системная терапия оправдана при умеренном или тяжелом СЛ. Признаки, которые могут указывать на среднетяжелое или тяжелое заболевание, включают глубокое вовлечение подкожной клетчатки, фасций и мышц, пересекающих сустав и потенциально вызывающих функциональные нарушения, линейные или атрофические поражения, поражающие лицо/кожу головы, быстро прогрессирующее или широко распространенное активное заболевание, а также неэффективна местная или УФ-терапия [86, 89].Наиболее часто используемой системной терапией является метотрексат (МТ) в сочетании с кортикостероидами (КС), что подтверждено недавним опросом 158 педиатрических ревматологов Северной Америки, проведенным Li et al. [89]. Клинические виньетки были розданы клиницистам, описывающим пациентов с умеренно-тяжелым LS, и клиницистов попросили дать свои рекомендации по лечению. Реакция клиницистов на начальную терапию заключалась в подавляющем большинстве случаев в комбинации с метотрексатом и кортикостероидами (более 80% использовали бы оба препарата). Несмотря на то, что было достигнуто отличное согласие в отношении выбора лекарств, схемы лечения широко различались в отношении приема (пероральный или комбинированный).подкожно или внутривенно), а также снижение дозы кортикостероидов и продолжительность терапии [89].
Подкомитет CARRA вырабатывает наилучшие согласованные протоколы лечения для пациентов с умеренным и тяжелым LS, чтобы помочь решить эту проблему и сузить список схем, которые были бы полезны клиницисту. Эти протоколы имеют четыре плана лечения для индукции терапии. Три протокола включают подкожное введение метотрексата (1 мг/кг, максимальная доза 25 мг в неделю), а четвертый был разработан для тех, у кого метотрексат неэффективен или у кого непереносимость метотрексата.Протоколы подразделяются на монотерапию метотрексатом, метотрексат с пероральным КС и постепенной дозировкой, метотрексат с внутривенным КС и микофенолата мофетил (ММФ) в дополнение к пероральному или внутривенному КС. Эти протоколы в настоящее время представляются для публикации. Было показано, что MMF эффективен при тяжелом резистентном к метотрексату педиатрическом LS, о чем свидетельствует серия случаев из 10 пациентов (два с пансклеротическим морфеозом, три с GM, два с LiScl конечностей и 3 с LiScl головы). Во всех случаях наблюдалось улучшение при приеме ММФ и удалось снизить дозу кортикостероидов [90].
Первое двойное слепое рандомизированное контролируемое исследование у детей с LS с использованием объективных показателей было проведено Zulian et al., в котором сравнивались метотрексат и пероральные кортикостероиды с метотрексатом и пероральными кортикостероидами.плацебо и пероральные КС. Было установлено, что метотрексат эффективен и безопасен, что подтверждает поддержку сообществом использования метотрексата при LS. Кроме того, у тех, кто лечился метотрексатом, значительно улучшилось состояние и было значительно меньше рецидивов по сравнению с теми, кто принимал только КС [84].
Другие методы лечения
Интенсивная физиотерапия и трудотерапия в сочетании с системной иммуносупрессивной терапией рекомендуется пациентам с линейной или глубокой склеродермией конечностей, чтобы помочь избежать или свести к минимуму контрактуры суставов.Кроме того, для пациентов с ECDS и / или PRS вмешательство пластической хирургии считается разумным после того, как заболевание находится в стадии ремиссии без каких-либо признаков клинической активности. Аугментация включает в себя растирание жировой ткани микрокаплями из подкожной клетчатки живота пациента, чтобы помочь заполнить часть подкожной атрофии, чтобы получить более симметричный контур лица (). При более тяжелом заболевании кожно-фасциальные лоскуты и реконструкция лицевых костей также являются вариантами, хотя обычно их применяют в позднем подростковом возрасте, после того как скелет созрел [91].Один центр сообщает, что из 10 подростков с ECDS и/или PRS, перенесших хирургическое вмешательство для коррекции лицевой деформации, 9 сообщили об удовлетворении результатами процедуры (процедур), и все они рекомендовали бы хирургическое вмешательство другим людям с этим заболеванием, что означает потенциальную большую пользу. влияние на качество жизни этих пациентов [92].
Синдром Парри-Ромберга, поражающий левое лицо до (А) и после (В) пересадки подкожно-жировой клетчатки.
Склеродермия у детей и подростков
Когда у вашего ребенка диагностируют склеродермию, это может быть чрезвычайно тревожным, поэтому важно иметь надлежащий уровень информации и поддержки.Мы всегда готовы предоставить любые ресурсы, которые могут вам понадобиться, и ответить на любые вопросы, поэтому звоните в нашу бесплатную службу поддержки по телефону 0800 311 2756.
Существуют различные виды склеродермии с различной степенью тяжести. При склеродермии у детей более легкие формы встречаются гораздо чаще, чем более серьезные.
Локализованная склеродермия
Большинству детей со склеродермией ставится диагноз «локализованная склеродермия». Это означает, что задействована только кожа; а остальная часть тела останется незатронутой.Существует несколько типов локализованной склеродермии, которые могут возникать у детей:
Морфея
Морфея является наиболее распространенной формой склеродермии у детей. Он проявляется в виде пятен неправильной формы на коже, которые сначала кажутся маленькими и розовыми, а затем затвердевают, что приводит к диагнозу склеродермия. Перспективы у детей с морфеей превосходны, потому что эти пятна со временем размягчаются и исчезают, не оставляя серьезных долгосрочных осложнений.
Линейная
Линейная склеродермия проявляется в виде линий натянутой, блестящей и часто потемневшей кожи. Они часто появляются на руке или ноге ребенка и распространяются на руки или ноги. Он также может поражать подлежащие мышцы, жир и кости, что приводит к рубцовому внешнему виду. Ранняя диагностика и лечение линейной склеродермии очень важны для растущих детей, особенно если пораженный участок кожи пересекает сустав, поскольку это может повлиять на нормальный рост и развитие.
Дети с линейной склеродермией часто имеют положительный прогноз, при условии, что они получают надлежащее и своевременное лечение для сведения к минимуму любых долгосрочных осложнений.
Линейная склеродермия с саблевым ударом
Саблезубый приступ возникает, когда у детей линейная склеродермия проявляется на лице и, возможно, на волосистой части головы. Если пораженный участок ограничен кожей головы и лбом, то проблема в основном косметическая.
Синдром Парри-Ромберга
Этот тип склеродермии характеризуется поражениями кожи, сходными с поражением кожи, но может поражать всю сторону лица ребенка и в некоторых случаях язык.При поражении одной стороны лица могут возникнуть дополнительные осложнения, поскольку лицевые кости могут неправильно расти.
Лечение локализованной склеродермии обычно включает противовоспалительные препараты, физиотерапию и трудотерапию.
Системный склероз у детей
У очень небольшого числа детей диагностируется «системный склероз», более серьезная форма склеродермии. При системной склеродермии поражаются как кожа, так и внутренние органы.Из всех детей с диагнозом склеродермия менее 10% будут иметь эту форму заболевания.
Системный склероз может появиться в любом возрасте. До полового созревания встречается в равной степени у мальчиков и девочек; однако после полового созревания чаще возникает у девочек.
Дети с системным склерозом обычно имеют синдром Рейно. Поражение внутренних органов может привести к таким симптомам, как затруднение глотания и изжога. Иногда могут быть поражены сердце, легкие или почки, что может потребовать специальных анализов и лечения.
Как и при локализованной склеродермии, лечение обычно включает медикаментозное лечение и физиотерапию, однако детям с системным склерозом потребуется квалифицированная медицинская помощь и долгосрочная поддержка во взрослом возрасте.
Лечение склеродермии у детей
Детская склеродермия 101
В качестве детского ревматолога в HSS я лечил несколько молодых пациентов с различными формами склеродермии. Хотя многие считают склеродермию болезнью взрослых, она может встречаться и у детей, и между обеими формами есть важные различия.Склеродермия у детей встречается редко, и могут пройти месяцы или даже годы, прежде чем пациенту будет поставлен правильный диагноз. Детский ревматолог лучше всего подходит для диагностики и лечения склеродермии у ребенка.
Склеродермия означает «твердая кожа».
- Название склеродермия представляет собой сочетание греческих слов «твердый» и «кожа». Для многих детей это основное проявление данного заболевания. Сначала кожа может казаться толстой или опухшей, а со временем может приобрести восковидную текстуру и стать стянутой или стянутой.У некоторых детей есть форма склеродермии, при которой изменяется кожа по всему телу, а у других форма, при которой поражается только небольшая часть.
Существуют две основные формы склеродермии у детей.
- Системный склероз: Дети с системным склерозом страдают заболеванием, поражающим многие системы органов в их организме. Этот тип склеродермии чаще встречается у взрослых и очень редко у детей. Первым симптомом у многих детей является феномен Рейно, при котором изменение цвета пальцев может произойти при воздействии холода.Эти дети могут испытывать такие симптомы, как усталость, мышечные боли или боли в суставах. У них также могут быть проблемы с желудочно-кишечным трактом, включая проблемы с глотанием, кислотный рефлюкс, запор или диарею. В какой-то момент у них могут развиться проблемы с сердцем, легкими или почками. В связи с этим важно, чтобы дети с системной склеродермией находились под тщательным наблюдением врача, имеющего опыт лечения этого заболевания.
- Локализованная склеродермия (морфеа): По сравнению с детьми с системным склерозом у детей с локализованной склеродермией, также известной как морфеа, заболевание, которое обычно поражает только их кожу.Хотя это все еще редкое заболевание, это наиболее распространенный тип склеродермии у детей и обычно не проявляется во взрослом возрасте. При самой легкой форме, ограниченной морфее, у детей может быть только одно или два небольших круглых поражения кожи, которые могут выглядеть как родимые пятна. У некоторых детей наблюдается огрубение кожи на части руки или ноги; если эта кожа пересекает сустав, это может вызвать проблемы с движением конечности. Локализованная склеродермия также может поражать лицо. Пациенты с наиболее тяжелой формой поражения лица, также известной как синдром Парри-Ромберга, также могут иметь поражение головного мозга или глаз.
Склеродермия лечится иммунодепрессантами.
- Лечение склеродермии зависит от типа заболевания и степени поражения кожи и внутренних органов. Детям с легкой ограниченной морфеей могут потребоваться только местные лекарства, обычно назначаемые детским дерматологом. Большинству других детей с локализованной склеродермией потребуется лечение более сильными препаратами. Как правило, при локализованной склеродермии используется лекарство, известное как метотрексат, которое можно принимать перорально или в виде инъекций.Также могут использоваться другие лекарства, включая стероиды. У некоторых пациентов локализованное поражение склеродермией становится неактивным в течение нескольких месяцев или лет, и в этот момент лекарство можно безопасно отменить при тщательном наблюдении. Детям с системным склерозом обычно требуется длительное лечение. Не существует единственной терапии, которая работала бы для всех педиатрических пациентов с системным склерозом, и лечение тщательно подбирается с учетом симптомов и вовлеченных систем органов. Лекарства могут включать антигипертензивные средства, такие как нифедипин, иммунодепрессанты, такие как метотрексат, и химиотерапевтические средства, такие как циклофосфамид.В дополнение к медикаментозному лечению пациенты со всеми формами склеродермии могут воспользоваться физиотерапией, группами поддержки и ранним обучением.
Исследования важны для лучшего понимания причин и методов лечения.
- В настоящее время причина склеродермии и взаимосвязь между различными типами до конца не изучены. Генетика, окружающая среда и аномалии в клетках кожи, кровеносных сосудов и иммунной системы, вероятно, играют определенную роль.В настоящее время в HSS ведутся исследования причин этого сложного заболевания. Кроме того, HSS сотрудничает с другими учреждениями по всей стране и миру, чтобы лучше понять течение, прогноз и оптимальное лечение детей со склеродермией.
Доктор Сара Табер является Детским ревматологом в Больнице специальной хирургии и специализируется на диагностике и лечении детей с ревматическими заболеваниями, включая ювенильный лювенит, идиопатический артрит у детей системная и локализованная склеродермия.
.