
Спинальная мышечная атрофия: причины, симптомы и прогноз
Страшно узнать, что малыш никогда не будет сидеть, стоять, бегать. Еще страшнее видеть, как нормально растущий и развивающийся ребенок вдруг начинает медленно угасать, постоянно падать, через несколько месяцев не может подняться по лестнице, а однажды теряет способность просто встать.
Спинальная мышечная атрофия
Врачи объединяют несколько видов наследственных заболеваний, характеризующихся нарушением движения, в одну группу под названием спинальная мышечная атрофия. В МКБ-10 они идут под кодом G12 с дополнительными указаниями на тип болезни.
По данным исследователей, около 0,01-0,02% детей рождаются с диагнозом СМА. Чаще патология встречается у мальчиков и мужчин.
Обнаруживается спинальная мышечная атрофия преимущественно у детей в раннем возрасте. Однако некоторые формы заболевания начинают проявляться только у подростков или уже взрослых людей. Коварство патологии заключается в том, что она постепенно, день за днем отбирает у больных то, что они сумели добиться.
Впервые патологию описал Г. Вердниг. Он обратил внимание на равностороннюю атрофию спинного мозга, его передних рогов, корешков периферических нервов в 1891 г. Уже в следующем году Дж. Хоффман сумел доказать, что речь идет о самостоятельном заболевании. В середине XX в. исследователи Е. Кугелберг и Л. Веландер описали патологию, которая возникает в позднем возрасте и имеет более благоприятный прогноз.
Симптомы
Каждый вид СМА имеет свои особенные признаки, однако существуют некоторые симптомы, которые позволяют объединить разнородные заболевания в одну группу. Это:
- Нарастающая слабость мышц и их атрофия.
- При заболевании, проявившемся после 1-2 лет, заметна деградация уже достигнутых способностей, например, бега, ходьбы.
- Тремор пальцев. Дрожь наблюдается и на языке.
- Деформация скелета.
- Сохранность интеллектуального и психического здоровья у большинства больных.
Виды СМА
Возраст, время проявления симптомов, особенности течения патологии, прогноз позволяют выделять несколько видов заболеваний.
СМА 0
Данная форма патологии описывается редко, часто его объединяют с первым типом СМА. Болезнь – врожденная. Характеризуется полным отсутствием движений, сухожильных рефлексов, слабостью мышц, ограниченным движением суставов коленей. С самого рождения наблюдаются дыхательные нарушения.
Часто диагноз путают с перинатальной энцефалопатией или родовыми травмами. Однако в последних двух случаях дети достаточно быстро адаптируются, их состояние становится лучше. У детей со СМА улучшения не возникают, в большинстве случаев они умирают, не дожив до месяца, от осложнений.
СМА-1
Патология первого типа имеет очень тяжелое течение. Ее называют также болезнью Верднига-Гоффмана. Диагностирован этот тип может быть от рождения до 6 месяцев. Отмечается слабость мышц, их периодическое подергивание – последнее увидеть достаточно трудно из-за достаточно большого слоя жирового слоя. Дрожь может периодически пробегать по языку малыша.
Наблюдается ухудшение рвотного, сосательного, глотательного рефлекса, нарушение слюноотделения. Младенец не может кашлять, громко кричать. Часто сопровождается тяжелыми дыхательными нарушениями, пневмонией.
Грудная клетка у таких детей имеет более плоскую форму из-за слабо развитых мышц груди.
Малышей со спинальной амиотрофией Верднига-Гоффмана легко узнать по позе лягушонка. Бедра и плечи отведены, локти и колени согнуты.
К 6 месяцам ребенок может научиться держать головку, но практически никогда не сможет самостоятельно сесть, встать, ходить. Проблемы с глотанием вызывают сложности в кормлении.
Часто именно это заболевание сопровождается олигофренией, врожденными нарушениями работы сердца, небольшим размером головы.
Поздняя младенческая
Патология второго типа обнаруживается у малышей в возрасте от полугода до полутора-двух лет. Болезнь Дубовица характеризуется слабостью и тремором в глубоких отделах мышц, дрожью пальцев, языка, ограничением объема движения конечностей. Детей отличает маленький вес, задержка развития. Они сидят, сами кушают, но вставать и ходить не могут.
Болезнь носит прогрессирующий характер. Со временем слабеют мышцы груди, шеи, исчезают сухожильные рефлексы, отмечаются нарушения глотания, слабый голос. Больного можно узнать по свисающей головке.
Ювенильная
Патологию Кугельберга-Веландера диагностируют часто после 2 лет. Она считается относительно легкой формой СМА, многие больные доживают до 30-40 лет. Человек стоит, однако дается ему это с трудом из-за очень слабых мышц. Происходит постепенная атрофия мышц.
Ребенок до 10-12 лет развивается нормально, потом начинает спотыкаться, падает, теряет способность заниматься спортом, бегать, выходить из дома, просто перемещаться без инвалидного кресла. Больного мучают периодические судороги конечностей. Развивается сильный сколиоз, изменяется форма грудной клетки.
Часто у таких пациентов происходят переломы, отмечается ограниченный объем движения суставов.
Поздние патологии
К четвертому типу относят бульбоспинальную амиотрофию Кеннеди, дистальную амиотрофию Дюшенна-Арана, а также перонеальную амиотрофию Вюльпиана. Заболевания обычно диагностируются в возрасте 35-40 лет, иногда возрастные границы расширяются от 16 до 60 лет. Больной отмечает постепенную потерю мышечной силы, угасание рефлексов сухожилий, видимые сокращения мышц.
При атрофии Дюшенна-Арана прежде всего поражаются кисти рук. Амиотрофию Вюльпиана можно узнать по формированию крыловидных лопаток.
Причины и механизм развития заболевания
Спинальная амиотрофия развивается из-за мутировавшего SMN гена пятой хромосомы. Если оба родителя – его носители, существует 25%-ная вероятность, что ребенок родится больным.
Мутация гена SMN приводит к нарушению синтеза белка, в результате чего происходит разрушение мотонейронов спинного мозга. Нервные импульсы не проходят к мышцам, которые из-за бездействия атрофируются, человек теряет способность двигаться.
Считается, что теряет работоспособность сначала глубоко расположенная мускульная ткань.
Диагностика
Наиболее точным методом определения спинально-мышечной атрофии у детей является анализ ДНК. Он проводится как у родившегося малыша, так и во время внутриутробного развития. Дополнительно проводятся следующие исследования:
- Анализ на биохимию. Целью является выяснение уровня ферментов: ананинаминотрансферазы, лактатдегидрогеназы, креатинкиназы. Нормальное их содержание позволяет исключить подозрения на прогрессирующую дистрофию мышц.
- Электрофизиологическое исследование. Метод направлен на регистрацию биоэлектрической активности. Патологию характеризует ритм «частокола».
- МРТ. Назначается для обнаружения признаков атрофии мышц.
- Микроскопия спинного мозга. Отмечаются признаки дегенеративных процессов в клетках нервных отростков. Они сморщиваются, разбухают, при этом глиальные волокна имеют плотную структуру.
- Тандемная масс-спектрометрия. Исследование помогает уточнить уровень аминокислот и белка СМН.
- Гистологическое исследование поперечнополосатых мышц. По результатам будут видны группы мелких волокон.

Если у молодых людей, планирующих рождение ребенка, есть родственники с патологией СМА, им рекомендовано пройти генетическую экспертизу.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
Витамины и витаминные комплексы:

Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Прогноз
То, как будет развиваться болезнь, сколько лет проживет ребенок, зависит от ее типа.
При атрофии типа один прогноз крайне неблагоприятен. Около 50% малышей не доживают и до двух лет. Не больше 10% детей с болезнью Верднига-Гоффмана могут дожить до пяти лет. Причиной гибели чаще всего становится воспаление легких, остановка дыхания, сердца.
Пациенты, которым диагностирована болезнь Дубовица, живут в среднем до 10, иногда 12 лет. Около 30% малышей умирают, не достигнув четырех лет.
При SMA III типа детская смертность встречается реже. У многих пациентов симптомы появляются в предподростковом-подростковом возрасте. Через несколько лет они перестают ходить. Далее, по нарастающей, отмечается атрофия мышц внутренних органов, в том числе дыхательных.
Считается, что заболевание IV типа не влияет на продолжительность жизни, тем не менее, оно ведет к инвалидизации.
Профилактика
Мер, направленных на профилактику и предотвращение развития СМА, не существует. Женщина, ожидающая рождения ребенка, может заподозрить проблему, обратив внимание на слабость шевелений плода. Проведенный ДНК-анализ может подтвердить или развеять подозрения. При необходимости проводится медицинская комиссия, которая может порекомендовать прерывание беременности. Врач обязательно рассказывает о заболевании, его течении и последствиях.
После диагностики заболевания у уже родившегося ребенка его окружают заботой и вниманием. Использование системы искусственной вентиляции легких, отсасывателей мокроты, специальных приспособлений для движения малыша, который может передвигаться, помогают улучшить качество жизни и помочь ребенку жить. Рекомендовано регулярно делать массаж, физиопроцедуры. Детей даже с ограниченными движениями возят в бассейн.
Спинальная амиотрофия – опасная, пока не поддающаяся лечению патология. Она характеризуется атрофией мышц. Возникает в разном возрасте. Прогноз в большинстве случаев неблагоприятный.
Для подготовки статьи использовались следующие источники:
Селиверстов Ю. А., Клюшников С. А., Иллариошкин С. Н. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения // Журнал Нервные болезни — 2015
Лепесова М. М., Ушакова Т. С., Мырзалиева Б. Д. Дифференциальная диагностика спинальной мышечной амиотрофии первого типа // Вестник Алматинского государственного института усовершенствования врачей — 2016
neuromed.online
Спинальная мышечная атрофия – виды, симптомы и возможности лечения
Спинальная мышечная атрофия является основной генетической причиной смерти в детском возрасте. Давайте разберемся в причинах и способах изменения жизни ребенка, а также узнаем, какие методы лечения доступны на сегодняшний день.
Что такое спинальная мышечная атрофия
Спинальная мышечная атрофия (SMA: Spinal Muscular Atrophy) – это нервно-мышечное заболевание аутосомно-рецессивного типа, характеризуется гибелью двигательных нейронов, расположенных в переднем роге серого вещества спинного мозга и в нижней части ствола головного мозга.
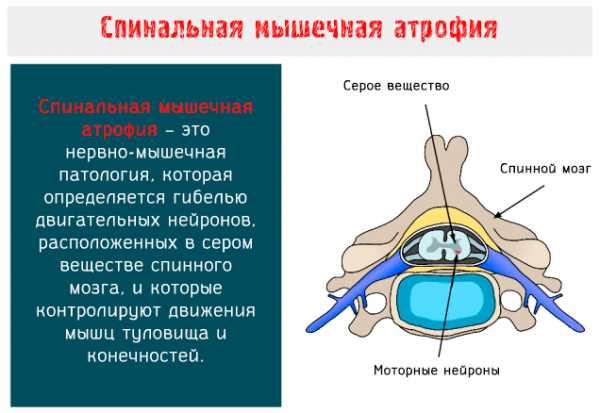
Моторные нейроны – это клетки, из которых образуются нервы, предназначенные для управления скелетными и поперечно-полосатыми мышцами глотки и гортани: когда они вырождаются, целые группы волокон подвергаются атрофии и, соответственно, результатом является мышечная слабость.
Работы глазных мышц, хотя и управляется моторными нейронами энцефального ствола мозга, не нарушается при этой болезни.
Частота спинальной мышечной атрофии колеблется от 1:6000 до 1:10000, и подвержены ей все этнические группы; является редким заболеванием, является одним из самых распространенных нервно-мышечных заболеваний, точнее вторым после дистрофии Дюшенна.
Причина спинально мышечной атрофии
Причина спинально мышечной атрофии была обнаружена в середине 90-х годов, спустя сто лет после первого описания болезни. В 95% случаев речь идёт о делеции в гене SMN1, локализованном на длинном плече хромосомы 5 (делеция – это потеря последовательности ДНК).
Поскольку спинально мышечная атрофия наследуется по аутосомно-рецессивному типу, для развития болезни человек должен получить обе копии плохого SMN1 – от матери и от отца. Таких родителей называют гетерозиготными или носителями, и они не имеют симптомов заболевания. Носители встречаются с частотой 1:50.
Ген SMN1 кодирует белок SMN, который используется в цитоплазме и ядре всех клеток и имеет решающее значение для формирования snRNP, малых ядерных рибонуклеопротеидов, компонентов сплайсинг машин.
Почему же вездесущий белок SMN является критическим фактором для выживания и надлежащего функционирования моторных нейронов?
В 2012 году Лотти и соавторы показали, что белки SMN имеют имеют важное значение для дифференциации и бесперебойной работы моторных нейронов.
Другие гипотезы были сформулированы для объяснения антиапоптической роли SMN:
- потребность в этом белке выше у моторных нейронов, чем в других тканях.
- по мнению других авторов, это можно объяснить тем, что белок SMN участвует в транспортировке вдоль аксонов РНК-связывающих белков.
Несмотря на все предположения, в настоящее время ещё неясно, какая из многих функций протеина SMN связана с развитием спинально мышечной атрофии.
4 типа спинально мышечной атрофии
Спинально мышечную атрофию классифицируют на четыре типа, в соответствии:
- с возрастом появления симптомов
- с максимальной двигательной активностью
У 25% лиц избегают точной классификации. Кроме того, у людей, страдающих от одного типа заболевания, симптомы могут существенно различаться.
1 тип – болезнь Верднига-Гоффмана
Это самая тяжелая форма спинально мышечной атрофии, составляет 50% от всех случаев.
Главные её особенности:
- проявляется до 6-го месяца жизни
- ребенок имеет плохую и дряблую мышечную массу: он мало движется, потому что не может противостоять силе тяжести, не в состоянии держать голову в вертикальном положении и сидеть без поддержки
- кости хрупкие и подвержены переломам, кроме того, в позвоночнике развивается сколиоз. Проблемы с костями у пациента со спинально мышечной атрофией не удивляют, так как именно физическая активность способствует минерализации костей
- рефлекс сосания и глотания слабый, поэтому такого ребёнка трудно кормить
- грудная клетка ребенка меньше нормы из-за слабости дыхательных мышц. Кашлевый рефлекс слабый, что нарушает процесс избавления от выделений (слизи и твердые частицы, включая микробов)
У детей, страдающих от спинально мышечной атрофии 1 типа, часто развивается пневмония, так как они не в состоянии избавиться от каких-либо патогенных микроорганизмов с кашлем, а также из-за потери контроля глотательных мышц, которые не могут предотвратить попадание слюны и кусочков пищи в легкие. Повторяющие пневмонии ведут, к сожалению, к дыхательной недостаточности.
Для тех, кто страдает от этой формой патологии прогноз неблагоприятный: смерть наступает в течение 2 лет, даже самое хорошее лечение продлевает жизнь только до 5 лет.
2 тип – болезнь Дубовица
Промежуточная форма спинальной мышечной атрофии.
Давайте посмотрим характеристики:
- проявляется между 6 и 18 месяцами
- ребенок показывает задержку в развитии моторики: не в состоянии сидеть, ему нужна поддержка, чтобы стоять, и никогда не научится ходить. Может иметь легкий тремор рук
- при этом типе также отмечается склонность к развитию сколиоза и хрупкости костей
- у некоторых маленьких пациентов дисфагия становится препятствием для поглощения достаточного для развития количества калорий
- кашлевый рефлекс может ослабнуть, облегчая возникновение респираторных инфекций
При спинальной мышечной атрофии 2 типа также высок риск развития дыхательной недостаточности. Прогрессирование симптомов настолько разнообразно, что некоторые пациенты умирают в младенчестве, другие в состоянии достичь зрелости.
3 тип – болезнь Кугельберга-Веландера
Детская форма спинальной мышечной атрофии, которая:
- может возникнуть в возрасте от полутора лет
- по сравнению с предыдущими случаями, дети могут стоять и ходить самостоятельно, эта способность в некоторых случаях сохраняется до зрелого возраста
- наблюдается тремор рук и могут возникнуть проблемы с суставами и сколиоз
- нарушения дыхания и глотания проявляются менее часто, чем при 1 и 2 типе
У людей, страдающих от 3 типа спинальной мышечной атрофии, средняя продолжительность жизни сравнима со здоровыми людьми. Но, из-за проблем с питанием и низкой физической активность, часто имеют избыточный вес.
4 тип спинальной мышечной атрофии
Это «взрослая» форма спинальной мышечной атрофии, более мягкое и менее распространенное заболевание.
Продолжительность жизни нормальная.
Как распознать спинальную мышечную атрофию
Специалист по детской неврологии задаст ряд вопросов, чтобы получить подробный отчет о медицинской истории ребенка и его семьи, после процедуры физического обследования, чтобы оценить физическое состояние маленького пациента.
Подтверждение диагноза спинальной мышечной атрофии достигается благодаря генетическому тесту: берут образец крови и исследуют на наличие аномального гена SMN1. Тест можно использовать, чтобы найти носителей.
Поиск неисправного SMN1 также может осуществляться путём биопсии ворсинок хориона, которые являются частью плаценты, что делает возможным пренатальную диагностику в случае:
- если у пары уже был ребёнок, пострадавший от спинальной мышечной атрофии
- партнеры обнаруживают, что являются носителями, но все равно хотя родить ребёнка
Иногда бывает, что нельзя точно утверждать, что это спинальная мышечная атрофия. Тогда используют другие тесты, которые помогают провести дифференциальный диагноз между спинальной атрофией и другими патологиями нервов и мышц:
- электромиография, которая измеряет электрическую активность мышц
- мышечная биопсия, то есть изучение образцов мышечной ткани
- оценка концентрации креатина киназы, фермента уровень которого повышается при повреждении мышц
Как облегчить симптомы спинальной атрофии
На данный момент не существует лекарств для лечения спинальной мышечной атрофии, поэтому пациенты могут воспользоваться только поддерживающим лечением.
Поддерживающая терапия
Базируется на трёх «краеугольных камнях»:
Для пациентов школьного возраста важно, чтобы они активно участвовали в школьных мероприятиях, потому что их физическая инвалидность никак не влияет на способности к обучению.
Физиотерапия
Физиотерапия необходима независимо от возраста человека. Упражнения позволят максимизировать амплитуду движений, чтобы предотвратить или замедлить потерю мелкой моторики. Дети со спинальной мышечной атрофией 1 и 2 типа получают огромную пользу от гимнастики в бассейне, поскольку вода помогает стимулировать всю мышечную массу.
Пациентам с 3 типом спинальной мышечной атрофии нужны ортопедические устройства (инвалидные коляски, параподы и т.д.), которые обеспечивают удобство и мобильность. Упражнения также важны, поскольку помогают предотвратить сколиоз, который усугубляет проблемы с дыханием и движениями.
Диетология
Каждый человек, страдающий от спинальной мышечной атрофии, должен иметь свой индивидуальный план питания, чтобы предотвратить последствия недостаточного или избыточного питания.
У тех детей, которые имеют большие трудности при грудном кормлении, пережевывании пищи и глотании, нужно принять меры, чтобы избежать таких осложнений, как аспирационная пневмония.
- Вы можете прибегнуть к использованию назогастрального зонда, который проходит через нос и доставляет пищу в желудок. Его относительно легко установить и снять, но он может протекать, тогда его следует заменить
- Другой вариант – гастростомия, то есть вывод трубки из желудка; является более простым в обслуживании, но процедура выполняется в операционной под наркозом.
Дыхание
Есть в этом случае для со спинальной мышечной атрофией существует три цели:
- пациенты и всех люди, которые вступают в контакт с ними, должны быть вакцинированы, например, против вируса гриппа, пневмококковой инфекции и бактерии коклюша, потому что инфекции дыхательных путей могут быть очень опасными для таких пациентов
- если кашлевый рефлекс слабым, это можно исправить с помощью специального устройства (Cough Assist): оно создает быстрое изменение давления снаружи и внутри легких, и быстрое прохождение воздуха по дыхательным путям, что имитирует кашель, освобождая дыхательные пути от секрета и микробов
- наконец, важна оценка дыхательной функции этих субъектов по степени сатурации кислорода в крови. Если количества кислорода меньше потребностей, стоит серьёзно рассмотреть идею использования механического респиратора. Изначально он используется в случае инфекции дыхательных путей и во время сна; с развитием атрфоии – весь день.
Возможные терапевтические стратегии
Открытие причины заболевания открыло для исследовательских групп большое направление для поиска методов лечения, направленных на максимально возможное замедление прогрессирования симптомов: повышение уровня белка SMN.
- Поскольку спинальная мышечная атрофия является моногенным заболеванием, это позволяет вмешаться в корень недуга, предоставляя пациентам функционирующий ген SMN1 (генная терапия)
- У лиц, страдающих от спинальной атрофии, но имеющих ген SMN2, можно увеличить экспрессию этого гена и заблокировать исключение экзона 7 во время сплайсинга незрелой мРНК.
В обоих случаях количество функционирующего белка SMN увеличивается.
AVXS-101 – экспериментальный препарат, разработанный биотехнологической компании Авексис, которому удалось достичь 1 этапа экспериментов на людях, при оценке безопасности лечения, и он начинает проверку эффективности.
Авексис сосредоточились на детях, страдающих от спинальной мышечной атрофии 1 типа, потому что это самый распространенный и смертельный тип заболевания.
AVXS-101 состоит из большого числа частиц адено-ассоциированного вируса серотипа 9, неспособного к репликации, но содержащего одну копию нормального гена SMN1.
Вводится в организм внутривенно, в состоянии преодолеть гематоэнцефалический барьер и достичь моторных нейронов.
Молекула ДНК, переносимая каждым вирусным вектором, производится в лаборатории. Она не изменяет ДНК пациента; содержит промотор, т.е. последовательность, которая способствует транскрипции ДНК в РНК, и гарантирует постоянное производство протеина SMN.
Анализ промежуточных данных, опубликованных Авексис в апреле 2016 года показывает, что:
- AVXS-101 хорошо переносится маленькими пациентами с атрофией 1 типа
- ни один ребенок не испытал «события»: событие – это смерть или использование механического респиратора в течение 16 часов каждый день в течение 2 недель подряд не связанного с острой респираторной инфекцией
- было отмечено улучшение двигательных навыков
- 100% из пациентов, которые не имели сложностей с кормлением, оставались стабильными
- 8 из 10 детей, которые вошли в исследование без проблем с дыханием, по-прежнему не нуждаются в поддержке
sekretizdorovya.ru
Спинальная мышечная атрофия: виды, причины и лечение
Спинальная мышечная атрофия (СМА), или амиотрофия, — это заболевание наследственного характера, которое сопровождается острыми нарушениями в активности нейронов мозга спинного и головного. Процессы затрагивают мотонейроны. Впервые заболевание описали в соответствии с медицинской картиной в XIX веке. Оно относится к группе генетических нарушений, обусловленных мутациями.
Специфичность мышечной атрофии заключается в том, что только один вид спинально патологии – первый – развивается у новорожденного в течение 1-2 месяца жизни. Остальные формы болезни дают о себе знать только во взрослом возрасте. Сложную форму спинальной атрофии и методы ее лечения изучают такие дисциплины, как генетика, неврология и педиатрия.
Распространенность нарушения
Существуют разные данные о том, как часто встречается спинально-мышечная атрофия у новорожденных детей. Плотность случаев напрямую связана с населенностью того или иного места на планете. Из-за того, что патологию часто обнаруживают только во взрослом возрасте, количество случаев после 20 лет больше, чем во младенчестве. Примерно 1 человек из 20 000 страдает одной из форм нарушения.
Факт! Среди младенцев тяжелые формы спинальной болезни встречаются в среднем 5-7 раз на 100 000 человек.
Наследственный фактор же проявляется далеко не у всех. Так, родители могут оказаться носителями мутировавшего гена. Но проявится он только у ребенка с вероятностью в 50-70%. Считается, что распространенность СМА среди носителей – 1 на 80 семей, или на 160 человек разного пола.
СМА – это одна из самых распространенных форм дегенеративных процессов наследственного характера у детей. Она занимает 2 место после муковисцидоза и считается причиной №1 среди наследственных болезней, ведущих к смерти ребенка до достижения им 15-18 лет.

Летальный исход возникает на фоне дыхательной недостаточности. Чем раньше спинальная патология проявляет себя, тем хуже будет прогноз. В среднем, дети с мышечно-спинальной атрофией доживают до 10-11 лет. При этом состояние интеллекта не оказывает влияния на прогресс спинальной амиотрофии.
Нарушение встречается чаще у мальчиков, чем у девочек, и протекает у них гораздо сложнее. На 1 пациента женского пола приходится 2 пациента – мужского. Но с 8 лет учащение среди девочек увеличивается.
Генетические факторы болезни
Спинальная мышечная атрофия появляется при наследовании рецессивного генома 5 хромосомы. Если оба человека, которые родили младенца, являются носителями СМА, то с вероятностью не менее 25% передают ген ребенку. В результате нарушается синтез белковых структур, разрушение моторных нейронов спинного мозга происходит в несколько раз быстрее, чем восстановление.
В период эмбрионального развития нервная система ребенка производит только половину от необходимого объема моторных нейронов. Со временем при СМА этот процесс сильно замедляется. После рождения из-за недостатка структур развивается спинальная атрофия.

Особенности функционирования нейронов
Активный мозг постоянно посылает импульсы в спинной мозг, а проводниками служат нервные клетки. Они доставляют сигналы в мышцы, в результате чего запускается их движение. Если этот процесс нарушен, то движение становится невозможным.
При спинально-мышечной атрофии двигательные нейроны ног, входящие в состав спинного мозга, работают некорректно. Они отвечают за сигналы, с помощью которых мозг поддерживает такие функции, как ползанье, поддержка шеи, сжимание и движение руками, ногами, а также дыхание и глотательный рефлекс.
Важно! При получении дефектных копий гена SMN1 от родителей, нервная система ребенка перестает производить белок, который контролирует процесс синтеза и обмена нейронов.
В результате мышцы, не получающие постоянных сигналов, начинают атрофироваться.
Классификация типов атрофии
Существует 4 распространенных группы спинально-мышечной атрофии у детей и взрослых:
- Младенческая форма. Самый сложный тип мышечно-спинальной атрофии, называемый также патологией Верднига-Гоффмана. Течение патологии при этой форме осложняется быстрым развитием тяжелых симптомов: появляются трудности с глотанием, сосанием и дыханием. Младенцы с СМА1 не могут нормально держать голову, сидеть.
- Промежуточная форма. СМА2, или болезнь Дубовица, несколько отличается степенью тяжести. При этой форме патологии ребенок может сохранять сидячее положение и даже есть, так как глотательные функции частично не нарушены. Но ходить он не сможет. Прогноз напрямую связан со степенью повреждения респираторных мышц, отвечающих за активность легких.
- Юношеская форма. СМА3, или болезнь Кюгельберга-Веландер, переносится подростками проще, чем первые типы спинально-мышечной атрофии. Ребенок может стоять, однако будет страдать от сильной слабости. Высок риск инвалидности – потребность в коляске сохраняется у большинства.
- Взрослый тип. СМА4 встречается в основном после 35 лет. Продолжительность жизни при заболевании не изменяется, но у пациента появляется выраженная слабость мускулатуры, снижение сухожильных рефлексов. По мере прогрессирования, требуется инвалидное кресло.

Заподозрить спинально-мышечную патологию сразу же после рождения очень сложно. Но обнаружение на первых этапах способно облегчить страдания пациентов, поэтому нужно знать о распространенных симптомах спинально-мышечной атрофии.
Симптомы разных форм болезни
Существует общий набор признаков СМА, по которым можно заподозрить патологию, если других проблем не обнаруживается, либо диагноз вызывает сомнение. Группу симптомов сводят к проявлению вялого периферического паралича:
- выраженная мышечная слабость или атрофия разных мышечных групп;
- сначала в процесс вовлекаются конечности – симметрично, ноги, а затем руки, постепенно втягивается и туловища;
- отсутствуют расстройства чувствительности и тазовые нарушения;
- наиболее выраженные проблемы затрагивают проксимальные или дистальные мышечные группы.
У пациентов появляются подергивания и фибрилляции – мерцательные аритмии.
Признаки СМА1
Заболевание Верднига-Гоффмана бывает 3 видов:
- Врожденная форма. Начинается в течение 1-6 месяца жизни, обладает тяжелыми симптомами. Обнаружить признаки можно во внутриутробном развитии – эмбрион будет мало двигаться. Гипотония наблюдается сразу же после рождения ребенка. Такие младенцы не держат голову, не могут сидеть. Постоянно находятся в позе лягушки с раздвинутыми конечностями. Сначала симптомы появляются в ногах, затем в руках, после этого страдает дыхательная мускулатура. Психическое развитие у таких детей медленное, они редко доживают до 2 лет.
- Ранняя спинальная мышечная атрофия. Первые признаки начинают беспокоить пациента до 1,5 лет, чаще всего – после какой-либо инфекции. Даже если раньше ребенок мог стоять и сидеть, теперь он утрачивает эти функции. Развиваются парезы, а затем поражаются дыхательные мышцы. Ребенок умирает, как правило, в результате затяжной пневмонии или дыхательной недостаточности в возрасте 3-5 лет.
- Поздняя форма. Патология встречается после 1,5 лет, двигательные возможности сохраняются у ребенка до 10 лет. Медленный прогресс симптомов приводит к дыхательной недостаточности и смерти в возрасте до 18 лет.
СМА1 – наиболее тяжелая форма патологии, готовиться всегда приходится к худшему исходу.
Признаки болезни Кугельберга-Веландера
Возникает в возрасте от 2 до 15 лет. Сначала в процесс вовлекаются нижние конечности, затем пояс таза, на последних стадиях страдает плечевой пояс и дыхательная система. Примерно у 25% пациентов появляется синдром псевдогипертрофии мышц, из-за чего патологию путают с мышечной болезнью Беккера.
Спинальная мышечная атрофия Кугельберга-Веландера не сопровождается костными деформациями, и пациенты способны сами себя обслуживать в течение долгих лет.

Амиотрофия Кеннеди
Эта патология входит во взрослую группу, болеют представители мужского пола после 30 лет. Женщины от патологии не страдают. Течение – умеренное, сначала поражаются ножные мышцы, последующие 10-20 лет пациент сохраняет привычный ритм жизни. Только после этого начинают страдать мускулы рук и головы. У многих пациентов со временем наступают эндокринные изменения: атрофия яичек, отсутствие либидо, сахарный диабет.

Дистальная СМА
Эта форма спинально-мышечной атрофии также развивается у взрослых пациентов после 20 лет. Ее второе название – СМА Дюшенна-Арана. Риск развития патологии сохраняется вплоть до 50 лет. Атрофия начинается в руках, вызывает синдром «когтистой лапы», потом переходит на крупные мышцы. Со временем появляются парезы мышц нижних конечностей, а туловище страдает редко. Прогноз у этой формы благоприятный, если не присоединяется торсионная дистония или болезнь Паркинсона.

СМА Вюльпиана
Скапуло-перонеальная форма спинально-мышечной атрофии, сопровождаемая симптомом «крылатых» лопаток. Появляется в среднем в 20-40 лет, позже встречается реже. Поражается плечевой пояс, а через некоторое время – руки и нижние конечности. При этой форме спинальной болезни двигательные функции у пациента сохраняются на 30-40 лет.

Способы диагностики патологии
Распознать спинально-мышечную атрофию со 100% гарантией можно только с помощью анализа ДНК на молекулярно-генетические факторы. С его помощью можно найти дефективный ген в 5 хромосоме.
Также применяют биохимический анализ для выявления состояния белка. Электрофизиологическое исследование мозга необходимо для определения активности импульсов и нервных стволов. МРТ и КТ назначают редко, так как эти методы не имеют высокой эффективности.
Методы лечения
Эффективных способов лечения спинально-мышечной атрофии не существует. Однако легкие стадии поддаются коррекции. С помощью физиотерапии, массажа и медикаментов можно поддерживать комфортное состояние ребенка. Во взрослом возрасте терапия более эффективна, так как эти формы атрофии не настолько тяжело переносятся.
Медикаменты
Для коррекции работы мышечных волокон и нервных импульсов используют препараты, улучшающие кровообращение и замедляющие разрушение нейронов:
- Антихолинэстераза. Средства подавляют активность фермента, расщепляющего ацетилхолин: «Прозерин», «Оксазил», «Сангвиритрин».
- Витамины и БАД. Используют антиоксиданты, карнитин, витамины группы B для поддержания обмена веществ и тонуса.
- Ноотропы. Улучшают работу нервной системы: «Ноотропил», «Кавитон», «Семакс».
- Средства для активации обмена веществ. В эту группу входят различные продукты: никотиновая кислота, «Актовегин», «Калия оротат».
Также важно поддерживать правильное питание ребенка, не допускать злоупотребления жирами и рафинированными продуктами.
Физиотерапия
Физиотерапевтические процедуры при спинально-мышечной атрофии улучшают тонус, кровообращение, обмен веществ, помогают снизить болезненные ощущения. Назначают: УВЧ, электрофорез, мануальные техники, дыхательные аппараты для стимуляции легких.
Внимательный контроль дыхания
Так как спинальная мышечная атрофия часто связана с такими нарушениями, как дыхание, необходимо строго следить за функционированием этой системы у ребенка:
- назначают физиотерапию грудной клетки;
- очищают дыхательные пути от образовывающейся слизи;
- назначают обезболивающие;
- принимают препараты, снижающие выработку секрета;
- используют методы неинвазивной вентиляции легких, которые повышают комфорт пациента и предотвращают гиповентиляцию ночью;
- применяют инвазивные методы – искусственную вентиляцию с помощью введенной трубки.
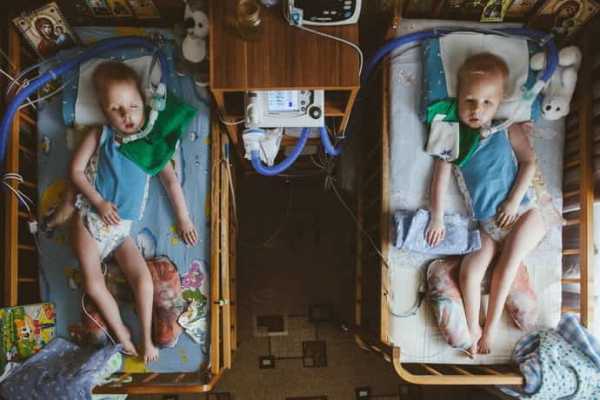
Последний метод используется в тяжелых случаях, когда дыхательный рефлекс становится невозможен.
Питание ребенка
Если спинальная мышечная атрофия развилась до такой степени, что пациент больше не может самостоятельно глотать, ему требуется сторонняя помощь. Нужно корректировать мышечную слабость.
Подробно о том, как кормить маленького пациента с нарушением глотательных функций, рассказывает доктор, ведущий мышечную атрофию. Иногда для реализации этих целей требуется профессиональная медицинская помощь.
Важно! При лечении больных с СМА не требуется соблюдение жесткого рациона или введения/ограничения каких-либо продуктов с содержанием определенных веществ, витаминов и минералов.
У детей с СМА может быть нарушен процесс пищеварения, из-за чего дети начинают мучиться от запоров. Иногда развивается рефлюкс-болезнь.
Прогноз и возможные последствия
Если спинальная мышечная атрофия обнаружена у пациента во взрослом возрасте, то прогноз более благоприятный. Патология СМА1 редко оставляет надежды – большая часть детей не доживает до 2 лет, остальные погибают в возрасте до 5 лет.
Смерть наступает из-за дыхательной недостаточности, реже – из-за острой, не проходящей, пневмонии. В настоящее время способов профилактики заболевания не существует.
Взрослые люди с диагнозом СМА должны отказаться от вредных привычек, экстремальных видов спорта и ненормированного режима отдыха/труда. Это значительно замедлит прогресс спинально-мышечной болезни.
nevrology.net
Типы СМА | Спинальная мышечная атрофия (СМА)
SMN-связанная СМА, или 5qSMA, или проксимальная СМА обычно подразделяется на три категории. Условно можно выделить еще СМА0 (ноль) и СМА4. Таким образом, есть несколько основных типов СМА, и все они протекают по-разному. Процесс может развиваться в различные периоды жизни, иметь свои клинические особенности, свой характер течения, прогноз и необходимый объем помощи и поддержки.
Тип 1 – наиболее тяжелый, с самым ранним дебютом, тип 3 – наименее тяжелый, с поздним возрастом начала. Некоторые специалисты выделяют еще тип 4 для обозначения умеренной или мягкой СМА с дебютом во взрослом возрасте.
Специфика заболевания в том, что у каждого ребенка, даже в пределах одной группы, СМА протекает по-разному, индивидуально. Внешне это проявляется в возможном объеме движений – некоторые дети способны удерживать голову, немного поднимать руки и приподнимать ноги, а другие просто лежат в классической позе «лягушка» и могут только чуть-чуть шевелить стопами и пальчиками кистей рук.
Виктор Дубовиц, один из корифеев в области СМА-ведения, в 90-е годы предложил помимо обычной классификации использовать более усложненную шкалу. Например, есть СМА1 и ее подтипы будут 1.1, 1.2, 1.3, т.е. от 1.1 до 1.9. Такая схема используется в Италии.

Виктор Дубовиц. Неразбериха в классификации СМА: возможность решения
Американская система основывается на шкале ABC, когда В – это классика, А – слабый тип, а С, соответственно, более сильный. Система с подтипами ABC наглядно описывает клинику СМА и позволяет подбирать для ребенка поддерживающую терапию в зависимости от подгруппы и соответствующих прогнозов. Этой системой начинают использоваться в России и других странах.
Важно! С помощью генетического теста тип СМА не определяется. Тип устанавливается на основе функциональных возможностей ребенка.
Симптоматика заболевания проявляется внутриутробно в отсутствии у плода двигательной активности. С рождения у ребенка выражена генерализованная мышечная гипотония с характерной позой «лягушки», резко снижена спонтанная двигательная активность. Сухожильные рефлексы не вызываются.
Как правило, эти дети долго наблюдаются педиатрами и неврологами с диагнозом перинатальная энцефалопатия. Иногда врачи связывают симптомокомплекс вялого ребенка с тяжелыми родами. Но все дети с перинатальной энцефалопатией и с последствиями тяжелых родов достаточно быстро и хорошо адаптируются, постепенно улучшаются, в отличие от детей со СМА.
Прогноз крайне неблагоприятный – дети погибают, как правило, в очень раннем возрасте (до шести месяцев) от интеркуррентных заболеваний (осложняющих течение основной болезни).
Обычно СМА0 и СМА1 объединяют.
СМА1
При I типе спинальной мышечной атрофии (тип Верднига-Гофмана) мать уже во время беременности может обратить внимание на позднее и слабое шевеление плода. С рождения у ребенка выражено распространенное снижение мышечного тонуса (синдром «вялого ребенка»). С первых месяцев жизни появляются слабость и атрофия мышц верхних и нижних конечностей, с последующим вовлечением мышц туловища и шеи. Такие изменения мышц приводят к тому, что дети не могут сидеть. Мышечные атрофии и подергивания мышечных волокон обычно маскируются хорошо выраженной подкожно-жировой клетчаткой. Характерным симптомом является мелкое дрожание (тремор) пальцев вытянутых ручек. Иногда обнаруживаются подергивания мышц языка.
Типичным признаком также является ослабление или полное исчезновение сухожильных рефлексов (коленный, ахиллов), ограничение нормальной подвижности в суставе, деформации скелета. Вследствие слабости межреберных мышц грудная клетка ребенка становится уплощенной. Так как в результате слабости мышц не происходит достаточной вентиляции легких, присоединяются частые респираторные инфекции, возникают разнообразные дыхательные расстройства. Психическое развитие детей не страдает.
У младенцев могут возникать дыхательные нарушения и невозможность приема пищи. Врожденные контрактуры, варьирующие по степени выраженности от простой косолапости до генерализованного артрогрипоза (множественная врожденная патология аппарата движения, проявляющаяся многочисленными контрактурами суставов, мышечной гипотрофией и поражением спинного мозга), возникают примерно у 10 % новорожденных с тяжелым поражением. Дети грудного возраста лежат в расслабленной позе «лягушка», спонтанная двигательная активность снижена, дети не могут преодолеть силу тяжести конечностей, плохо удерживают голову.
Обычно, за исключением очень редких, тяжелых случаев, диагноз СМА не ставится в роддоме. Ребенка выписывают домой здоровым, и когда родители замечают низкий мышечный тонус, они или не придают этому серьезного значения, если не знают, как должен двигаться здоровый малыш, или успокаиваются советами доктора в поликлинике: «Не переживайте, все развиваются в свой срок, еще встанет и побежит». Родители могут не понимать всей тяжести проблем, это должен увидеть врач, но, к сожалению, в поликлиниках симптоматику СМА знают плохо.
Эти дети проявляют первые признаки заболевания в возрасте до 6 месяцев. Это значит, что они не приобретают навыки самостоятельно сидеть, ползать, ходить. В итоге, объем движений у таких детей очень мал. При этом СМА не затрагивает когнитивную сферу, дети все понимают, не затронута и чувствительность. Если с ними заниматься, как с обычными детьми – играть, читать, собирать пирамидки, – то эти дети в умственном и психическом плане развиваются абсолютно нормально, и все задержки развития, которые могут им ставить неврологи, связаны с нарушением двигательной активности и условиями, в которых они находятся.
Более 2/3 детей с этим заболеванием умирают до 2-летнего возраста, во многих случаях смерть наступает в раннем младенческом возрасте в связи с поражением дыхательной мускулатуры и возникновением разнообразных осложнений со стороны легких.
СМА2
При спинальной мышечной атрофии II типа заболевание впервые проявляется несколько позже (в первые 1,5 года жизни ребенка) и характеризуется более медленным течением. Главным признаком является невозможность ребенка встать на ноги.
Дети со СМА2 обычно способны сосать и глотать, дыхательная функция в раннем младенческом возрасте не нарушена. Носовой оттенок голоса и нарушение глотания появляются в более старшем возрасте. Несмотря на прогрессирующую мышечную слабость, многие из них доживают до школьного и более старшего возраста, хотя на поздних стадиях заболевания отмечается тяжелая степень инвалидизации, и дети нуждаются в инвалидных колясках. Обычные коляски им не подходят, необходимы дополнительные поддержки, упоры, специальные приспособления, оптимально фиксирующие положение тела.
У многих пациентов с большой продолжительностью жизни одними из основных осложнений заболевания становятся сколиоз и контрактуры, которые развиваются очень быстро. Даже у лежачих детей сколиоз развивается достаточно существенно, искривление позвоночника происходит не от нагрузки, а от слабости.
Дети со СМА2 некоторый период времени бывают достаточно стабильны, и родители могут поддерживать тот набор навыков, который этими детьми уже приобретен.
Есть такой термин «плато заболевания», который обозначает некий период, в течение которого дети достаточно стабильны, и значительного ухудшения состояния, прогрессирования болезни не происходит. Это может выглядеть так: дети набирают навыки, как все обычные дети, при «запуске» болезни перестают развиваться, а потом регресс состояния у них может идти очень медленно или, наоборот, очень быстро, у каждого ребенка по-своему. Или так: дети определенное время набирают навыки, потом происходит какое-то событие или болезнь, часть навыков теряется, и дальше наступает довольно долгое «плато», в течение которого могут быть даже незначительные улучшения, но потом наступают неизбежные ухудшения. Скорость прогрессирования болезни, продолжительность «плато» (или его отсутствие) и последующих ухудшений – все это очень индивидуально.
СМА3
Болезнь Кугельберга-Веландер – наиболее легкая форма спинальной мышечной атрофии (СМА III типа). Начинается она в возрасте от 1,5 до 17 лет. С такой болезнью люди живут дольше, прогрессия идет медленно. Атрофия мышц начинается с ног и далее распространяется на руки. В младенческом возрасте клинические проявления заболевания могут отсутствовать. Прогрессирующая слабость развивается в проксимальных отделах конечностей, особенно в мышцах плечевого пояса. Пациенты сохраняют способность к самостоятельной ходьбе. Симптомы слабости мышц бульбарной группы появляются редко. Примерно у 25 % пациентов с этой формой СМА в большей степени выражена мышечная гипертрофия, а не атрофия мышц; поэтому может быть ошибочно диагностирована мышечная дистрофия. Пациенты могут дожить до зрелого возраста.
СМА у этой большой группы детей выявляется в возрасте старше полутора лет, т.е. обычно ребенок может самостоятельно ходить. Болезнь проявляется настолько индивидуально, что диагноз могут поставить в полтора года, а могут и в девять лет. В зависимости от этого будут разные прогнозы и по продолжительности, и по качеству жизни.
У детей со СМА3 продолжительность жизни почти не отличается от стандартной, они доживают и до 30, и до 40 лет. У них не так быстро прогрессируют все симптомы, но с этими детьми тоже надо проводить и реабилитационные занятия, и занятий по физической терапии.
Сейчас есть достаточно большое количество взрослых пациентов со СМА, и это не четвертый тип, это дети со СМА2 и СМА3, которые выросли. У них возникают большие проблемы, которые не связаны с перемещением и дыханием. Сколько бы они ни жили, 20 лет или 30, они уже разочаровались в медицине, потому что никто не знает, что с ними делать. Каждая мама ребенка со СМА и каждый взрослый пациент со СМА может рассказать примерно одну и ту же историю про прием у доктора: «Не приходите ко мне, мы не знаем, что с вами делать»; «Ты еще не умер, ах, ничего себе». Действительно, врачи не знают, что делать с этими пациентами, и говорят: «Ну, не лечится же, что ты хочешь от меня, правда, по-честному, я не знаю, что с тобой делать». Ими никто не занимается, после 18 лет вообще не знают, что с ними делать.
Эти пациенты (часто невидимые пациенты, так как сидят дома) не верят в медицинскую помощь, потому что всю жизнь от них отмахивались. И они обращаются за помощью только тогда, когда уже невозможно игнорировать симптоматику, когда сильная боль, т.е. поздно – практически уже на смертном одре. До недавнего времени с этой категорией пациентов не было никакой работы. Сейчас появилась возможность как-то улучшать ситуацию, больше помощи оказывают благотворительные фонды, этой проблеме уделяется больше внимания.
Заболевание СМА3 развивается не очень стремительно, постепенно происходит общее ослабление организма и медленная потеря приобретенных навыков.
СМА4
Четвертый тип СМА проявляется у людей старше 25 лет. Заболевание развивается довольно медленно, практически не влияя на продолжительность жизни. При СМА4 может развиваться тремор, возникать общее ослабление, в том числе, и мышечной силы. Со временем СМА4 может приводить к потере способности к самостоятельному передвижению.
spravka.neinvalid.ru
Самое частое из редких: что нужно знать о СМА
С помощью фонда «Семьи СМА» мы собрали главные факты об этой болезни, которые нужно знать

Фото с сайта hellohope.com
Частота заболевания
СМА – одно из самых часто встречеющихся заболеваний из орфанных (редких), болеет один новорожденный на 6000-10000.
Причина СМА
СМА — наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Если вашему ребенку диагностировали СМА, я хочу поговорить с вами
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.
Изображение с сайта bfm.my
От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III, болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА IV, этот тип называется ещё «взрослой СМА», поскольку болезнь проявляется обычно в возрасте после 35 лет.
Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.
Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук. Обычно проблем с глотательной и дыхательной функцией у больных нет.
Большинство из больных IV типом СМА могут ходить, и лишь некоторым приходится прибегать к инвалидным коляскам.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
Изображение с сайта asesoramientopsicologicoalicante.es
На сегодняшний момент радикального лечения от СМА не существует.
Международной корпорацией «Биоген» был разработан препарат «Спинраза», который значительно улучшил состояние больных, к которым применялся во время тестирования. В настоящее время препарат одобрен к применению в США, в Европе ориентировочная стоимость годового курса, по подсчётам компании, будет составлять около 270 тысяч евро, в России препарат не сертифицирован. Лечение пожизненное.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.

Фото с сайта f-sma.ru
Благотворительный фонд «Семьи СМА» помогает детям и взрослым со спинальной мышечной атрофией и другими нервно-мышечными заболеваниями и их семьям.Фонд работает по всей территории России. Работа фонда имеет два основных направления — оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования — например, СМА 300.
Ольга Германенко: «Болезнь дочери воспринимаю как награду»
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно. В этом отношении СМА – такая же «отложенная» болезнь как, например, миодистрофия Дюшенна или синдром Ретта, когда ребёнок, некоторое время развивавшийся в соответствии с нормой, теряет приобретённые ранее навыки и становится инвалидом.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
Сколько больных СМА в России?
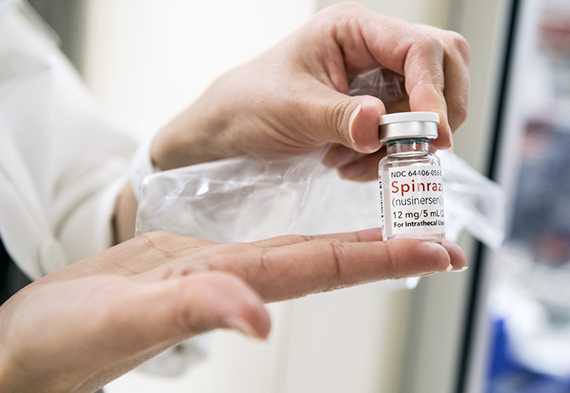
Препарат «Спинраза», значительно улучшающий состояние больных. Фото с сайта healthbeat.spectrumhealth.org
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Кто в России помогает людям со СМА и их семьям
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Известные люди со СМА

Валерий Спиридонов. Фото с сайта mioby.ru
Итальянка Симона Спиноглио родилась с наследственным заболеванием – спинальной мышечной атрофией 2 типа. Она с самого рождения не может ходить и передвигается только с помощью электрической коляски. Но ее жизнь полна и насыщенна; ничто не может помешать ее стремлению жить.
Симона работает на «горячей линии» итальянской Ассоциации «Семьи СМА» (Famiglies of SMA) и помогает детям и взрослым со СМА и другими нервно-мышечными заболеваниями.
Также Симона записала несколько популярных в итальянском сообществе СМА песен — о свободе делать то, что ты хочешь, несмотря на болезнь.
Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
В 2008 году собрала собственную музыкальную группу (распалась в 2010). В 2013 году приняла участие в конкурсе «Фактор А» на телеканале «Россия». Заняла второе место и получила персональную премию Аллы Пугачёвой «Золотая звезда Аллы». В 2017 году из-за недопуска России в конкурсную программу не смогла принять участие в конкурсе «Евровидение». Передвигается на коляске.
Программист из Владимира Валерий Спиридонов. Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Сегодня Валерий — член городской общественной палаты Владимира, эксперт по вопросам доступной среды, а также создатель собственного сообщества «Desire for life», рассказывающего о создании доступной среды и перспективных медицинских проектах. Валерий – участник многих телепрограмм на российском и зарубежном ТВ.
Валерий Спиридонов: «Мне сложно искать с Богом общий язык»
Полезные ресурсы про СМА
Сайт фонда «Семьи СМА» Фонд публикует информацию о болезни и свежие новости о лечении СМА в России. Много новостей выходит на странице фонда в ФБ.Американская ассоциация мышечных дистрофий (англоязычный),
Форум о миопатиях (российский).
Сайт о миопатиях (Беларусь):
Фонд «Дети со СМА» (Украина) (на сайте есть форум).
Благодарим за предоставленную информацию фонд «Семьи СМА» и лично Ирину Старову-Кислину.
www.miloserdie.ru
симптомы, лечение, особенности заболевания у детей
Каждому человеку крайне важно сохранять свою самостоятельность и активность. Однако существуют заболевания, при которых пациенты постепенно становятся недееспособными и могут передвигаться только с посторонней помощью или на инвалидных креслах. К таким типам болезней относится спинальная мышечная атрофия, при которой человек может не только перестать ходить, но даже порой оказывается не в состоянии самостоятельно дышать.
Описание болезни
Спинальная мышечная атрофия (СМА, спинальная амиотрофия) — это целая группа наследственных заболеваний, характеризующихся дегенерацией двигательных нейронов спинного мозга.
Это одна из самых распространённых патологий среди генетических нарушений. Частота встречаемости среди новорождённых — один случай на 6000–10000 детей, в зависимости от исследуемой страны. Практически половина родившихся с СМА не могут достичь даже двухлетнего возраста и погибают.
Однако мышечная атрофия не является только детским заболеванием, ей могут страдать люди разного возраста. Учёными было выяснено, что каждый 50-й житель Земли является носителем рецессивного гена SMN1 (survival motor neuron), приводящего к СМА. Хотя все виды этой болезни происходят от мутации в одном хромосомном участке, у неё существует множество форм с различными степенями проявления симптомов во всех возрастах. Несмотря на потерю двигательной активности мышц, их чувствительность сохраняется. Интеллект пациентов процессом не затрагивается, он полностью соответствует норме.
Впервые эта болезнь была описана в 1891 году Гвидо Вердингом, который не только зафиксировал симптоматику, но и изучил морфологические изменения в мышцах, нервах и спинном мозге.
Чаще всего встречаются проксимальные виды СМА (примерно 80–90% всех случаев), при которых сильнее поражаются мышцы, расположенные ближе к центру туловища (бедренные, позвоночные, межрёберные и т. д.).
Видео о пациенте со спинальной мышечной атрофией
Типы заболевания и степени выраженности проявлений
Среди проксимальных видов выделяют четыре формы заболевания, которые сгруппированы исходя из возраста начала процесса, тяжести проявления симптомов и средней продолжительности жизни.
Формы проксимальной спинальной амиотрофии — таблица
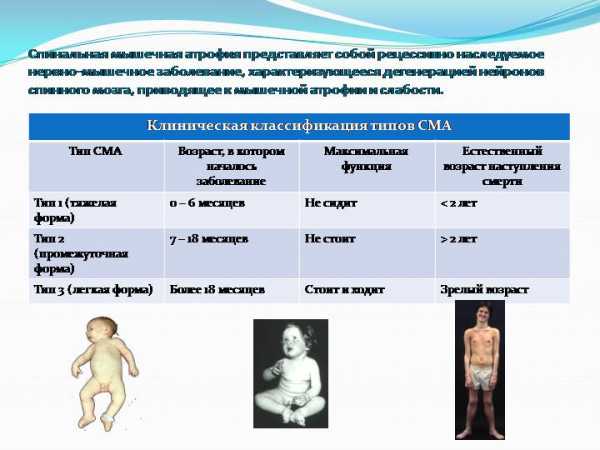
| Тип | Синонимы, другие обозначения заболевания | Средний возраст начала заболевания | Описание |
| I тип. Тяжёлая младенческая форма. | Болезнь Вердинга — Гоффмана | 0–6 месяцев |
|
| II тип. Поздняя младенческая форма. | Болезнь Дубовица | 6–18 месяцев |
|
| III тип. Ювенильная форма. |
| 2 года |
|
| IV тип. Взрослая форма. |
| 35 лет (но может встречаться с 16–60 лет) |
|
Существует ряд спинально-мышечных атрофий, задействующих дистальные отделы организма. Чаще всего поражаются верхние конечности, а сама болезнь обычно регистрируется в довольно взрослом возрасте.
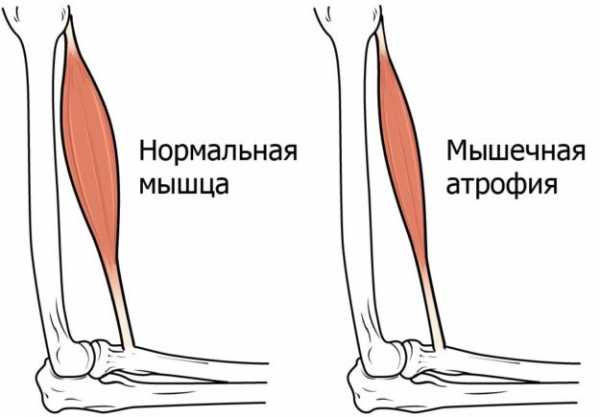
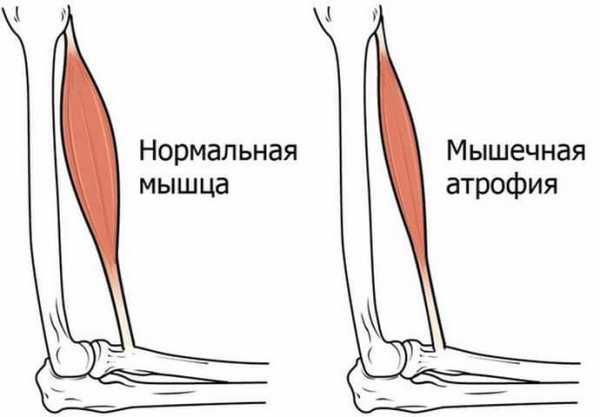
При СМА мышцы значительно утрачивают свою массу и объём
Помимо данной классификации, существует подразделение СМА на изолированные (происходящие только из-за поражения двигательных нейронов в спинном мозге) и сочетанные формы (к спинальной амиотрофии присоединяются такие дополнительные заболевания, как порок сердца, олигофрения, врождённые переломы и т.д.).
Причины и факторы возникновения патологии
Спинальные мышечные дистрофии возникают из-за наследуемых рецессивных генов, находящихся на пятой хромосоме (SMN, NAIP, h5F5, ВTF2p44). Как правило, у родителей не наблюдаются проявления СМА, но оба являются носителями и в 25% случаев передают дефектный ген своему ребёнку, который нарушает синтез белка SMN, что приводит к дегенерации моторных нейронов в спинном мозге.

Нарушения в работе всего лишь одного участка хромосомы приводят к различным видам СМА
На определённом этапе эмбрионального развития существует программируемая клеточная гибель предшественников двигательных нейронов, сформировавшихся в избытке. Из них в норме должна остаться примерно половина, которая в дальнейшем дифференцируется в нервные клетки. Однако в определённый период этот процесс останавливается с помощью функционирования гена SMN. При мутации его работа нарушается, гибель клеток продолжается даже после рождения ребёнка, что приводит к спинальной мышечной атрофии.
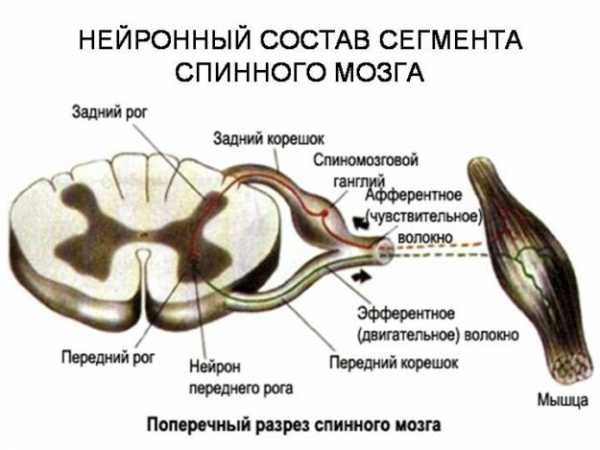
Заболевание поражает моторные нейроны передних рогов спинного мозга
Симптомы у ребёнка и взрослого
Основным признаком болезни СМА является мышечная вялость, слабость и атрофия. Однако у каждой из форм спинальных амиотрофий существуют свои особенности:
- При заболевании Вердинга — Гоффмана первые симптомы могут быть обнаружены ещё во время беременности на УЗИ осмотре, так как плод очень слабо шевелится. После родов отмечается невозможность ребёнка самостоятельно держать голову, переворачиваться и позднее сидеть. Почти всё время малыш лежит в расслабленной позе на спине, не имея возможности свести ноги и руки. Также отмечаются частые проблемы с кормлением, так как младенец испытывает трудности с глотанием. Дыхание зачастую нарушено из-за атрофии рёберной мускулатуры. Практически 70% детей погибают, не дожив до двух лет. После диагностики выявляется недостаточная сформированность передних рогов спинного мозга. Если пациент доживает до 7–10 лет, то у него нарастает выраженность мышечной атрофии и он погибает от острой сердечной, лёгочной недостаточности или из-за проблем с пищеварением. В редких случаях больные доживают до 30 лет, и то исключительно при более позднем начале проявления симптомов (около 2 лет).
- При втором типе спинальной мышечной атрофии ребёнок зачастую может самостоятельно дышать и глотать пищу. Однако со временем происходит прогрессирование процесса, и в более старшем возрасте дети оказываются прикованными к инвалидным креслам. Обычно родители начинают замечать, что ребёнок часто спотыкается, падает и у него подгибаются колени. Постепенная невозможность самостоятельно проглатывать пищу появляется с возрастом. Также по мере взросления начинает проявляться сильно выраженное искривление позвоночника (сколиоз). Эта форма считается относительно доброкачественной и позволяет пациентам прожить до старости. В некоторых случаях женщины даже могут выносить и родить ребёнка, однако велик шанс передачи болезни по наследству. При правильном уходе и благодаря регулярным занятиям лечебной физкультурой пациенты могут очень долгое время сохранять дееспособность.
- Ювенильная амиотрофия Кюгельберга — Веландера может впервые регистрироваться в возрасте от двух до восемнадцати лет. На самом раннем этапе симптомы могут отсутствовать, ребёнок полноценно развивается. Постепенно начинает появляться слабость в проксимальных отделах тела, чаще всего в плечах и предплечье. В течение многих лет пациент способен самостоятельно передвигаться и обслуживать себя. Часто наблюдаются мышечные подёргивания (фасцикуляции). Основной пик проявления симптомов регистрируется в возрасте от двух до пяти лет, когда ребёнку вдруг становится сложно бегать, вставать с кровати и подниматься по лестнице. Течение болезни относительно доброкачественное, так как пациент может длительно сохранять возможность самостоятельно передвигаться.
- Бульбоспинальная мышечная атрофия Кеннеди — заболевание, сцепленное с полом, передаётся с Х хромосомой и проявляется исключительно у мужчин во взрослом возрасте. Прогрессирует болезнь медленно и начинается со слабости в мышцах бёдер, затем через 10–15 лет постепенно присоединяются бульбарные расстройства (поражения черепных нервов: языкоглоточного, блуждающего и подъязычного). Так как течение заболевания крайне медленное, то важные функции практически не успевают нарушаться и продолжительность жизни сильно не сокращается. Очень часто болезнь сопровождается эндокринными патологиями: атрофией яичек, снижением либидо, сахарным диабетом.
- Дистальная СМА Дюшена — Арана обычно регистрируется в возрасте 18–20 лет. Первыми поражаются кисти рук, затем полностью верхние конечности. В течение длительного времени постепенно наступает атрофия мышц ног. В крайне редких случаях заболевание останавливается на парезе одной из рук.
- Скапуло-перонеальная спинально-мышечная атрофия Вюльпиана впервые дает о себе знать в старшем возрасте (20–40 лет). Проявляется постепенной атрофией мышц плечевого пояса и разгибателей стопы и голени. Прогноз относительно благоприятный, так как, даже спустя 30 лет с момента начала заболевания, у пациента сохраняется возможность передвигаться самостоятельно.
СМА у беременных связана со множеством осложнений. Зачастую самостоятельно родить женщина не может и ей назначают кесарево сечение.
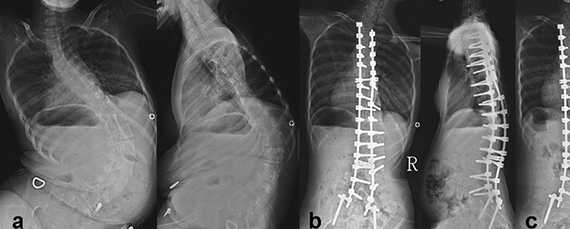
На рентгеновских снимках видно искривление позвоночника и последующее его исправление с помощью операции
Диагностика и дифференциальная диагностика
Метод, со 100% вероятностью указывающий на наличие спинальной мышечной атрофии — это анализ ДНК с помощью молекулярно-генетической диагностики. Он направлен на выявление дефектного гена в пятой хромосоме, в локусе 5q11-q13.

Анализ ДНК поможет с точностью установить диагноз
Биохимический анализ проводится с целью выявления содержания креатинкиназы (КФК), аланинаминотрансферазы (АЛТ) и лактатдегидрогеназы (ЛДГ). Если их уровень в норме, то это позволяет исключить схожую по симптомам прогрессирующую мышечную дистрофию.
С помощью ЭФИ (электрофизиологического исследования) регистрируются импульсы биоэлектрической активности мозга и нервных стволов. При СМА наблюдается характерный для поражения нейронов передних рогов ритм «частокола».
МРТ (магнитно-резонансная томография) и КТ (компьютерная томография) не всегда помогают выявить на изображениях характерные изменения для СМА.
Дифференциальная диагностика необходима для того, чтобы отличить спинальную мышечную атрофию от мышечной дистрофии, детского церебрального паралича, бокового амиотрофического склероза, синдрома Марфана, клещевого энцефалита и других заболеваний центральной нервной системы.
Тандемная масс-спектрометрия, позволяющая определить снижение уровня различных аминокислот в организме, выявляет недостаток белка SMN.
Лечение
На данный момент эффективного лечения спинальной мышечной атрофии ещё не придумано. При первичном обнаружении болезни показана обязательная госпитализация с проведением различных исследований.
Медикаментозная терапия
Лекарственная терапия направлена на улучшение проведения нервных импульсов, нормализацию кровообращения и замедление разрушения двигательных нейронов. Используют следующие препараты:
- антихолинэстеразные препараты направлены на снижение активности фермента, расщепляющего ацетилхолин, который, в свою очередь, передаёт возбуждение по нервным волокнам. К ним относятся Сангвиритрин, Оксазил, Прозерин;

Прозерин улучшает прохождение импульса от нейрона к мышце
- биологические добавки с содержанием L-карнитина и коэнзима Q10, усиливающие энергетический обмен в клетках;
- витамины группы В, поддерживающие нормальный мышечный тонус;
- ноотропы, стимулирующие работу центральной нервной системы — Ноотропил, Кавинтон, Семакс.
- препараты для стимуляции обмена веществ в мышечных и нервных волокнах — Калия оротат, Актовегин, Никотиновая кислота.
Актовегин улучшает метаболизм в нервной ткани

Диета
Следует понимать, что основу питания пациента с СМА должна составлять пища, которая может максимально обеспечить мышцы необходимыми веществами.
Стоит обогатить рацион больного продуктами с высоким содержанием белка. Однако достоверных данных, говорящих об улучшении состояния пациентов при определённом питании, в настоящее время нет. В некоторых случаях избыточное поступление аминокислот в организм может навредить, так как отсутствует достаточный объём мышечной ткани, способный их переработать.

Бобовые культуры — источник белка
Следует снизить калорийности пищи, так как из-за недостаточной двигательной активности некоторые пациенты склонны набирать лишний вес.
Физиотерапевтические методы, в том числе массаж
Больным необходимо проводить сеансы лечебного массажа, направленные на поддержание мышечных функций. Также полезным будет УВЧ (ультравысокочастотная терапия), электрофорез, мануальные практики. Существуют специальные дыхательные упражнения для стимуляции работы лёгких.

Массаж — средство лечения спинальной амиотрофии
С помощью нормированной физической активности можно предотвратить тугоподвижность суставов и держать мышцы в тонусе. Очень полезны занятия в бассейне, где идёт минимальная нагрузка на позвоночник. Важно подобрать правильные ортопедические приспособления, которые окажут поддержку грудной клетке и конечностям.

Больные СМА вынуждены пользоваться специальными ходунками, поддерживающими их во время ходьбы
В случае осложнений с питанием больным рекомендовано кормление через сформированное соустье между желудком и внешней средой (гастростому). При проблемах с дыханием назначается искусственная вентиляция лёгких.
Народные средства
Народных средств для лечения спинальной мышечной атрофии не существует. Очень важно сразу обратиться к врачу после обнаружения первых симптомов. Ни в коем случае нельзя заниматься самолечением, так как это может привести к летальному исходу.
Прогноз лечения и возможные осложнения
Прогноз лечения сильно зависит от типа спинальной мышечной атрофии. При самом злокачественном первом варианте наиболее частым исходом является ранняя смерть пациента, а в других случаях возможно порой до старости сохранять способность самостоятельно передвигаться.
К осложнениям при СМА относятся: сколиоз, острая лёгочная недостаточность, параличи, деформации грудной клетки, угасание жевательных и глотательных функций.
Профилактика
Профилактики спинальной мышечной атрофии не существует. Единственное, что можно предпринять — консультироваться у генетика во время планирования беременности.
Несмотря на страшные прогнозы и невозможность полностью вылечиться от этого заболевания, не стоит опускать руки. Полноценный уход и соблюдение всех рекомендаций врача во многих случаях позволяют пациентам прожить долгую и самостоятельную жизнь.
Высшее образование в сфере биологии (СПбГУ, магистр биологии), специализация — генетика человека. Оцените статью: Поделитесь с друзьями!Похожие статьи
spina-sustav.ru
Спинальная мышечная атрофия — это… Что такое Спинальная мышечная атрофия? 💯 ✅
Запрос СМА перенаправляется сюда. О стиральных машинах см. Стиральная машина
Спина́льная мы́шечная атрофи́я (СМА или SMA) — разнородная группа наследственных заболеваний, протекающих с поражением / потерей моторных нейронов передних рогов спинного мозга.
Для спинальных мышечных атрофий характерно нарушение работы поперечнополосатой мускулатуры нижних конечностей, а также головы и шеи. У больных отмечаются нарушения произвольных движений — ползание, ходьба, удержание головы, глотание. Мышцы верхних конечностей обычно не страдают. Для спинальных амиотрофий характерно сохранение чувствительности, а также отсутствие задержки психического развития.
Общая информация
- SMA (Спинальные мышечные атрофии) — одно из наиболее распространенных генетических нарушений (несмотря на редкую встречаемость),
- Спинальные мышечные атрофии детского возраста наследуются по аутосомно-рецессивному типу,
- Ген спинальной мышечной атрофии картирован на хромосоме 5 q11 .2 — 13.3,
- Этот ген СМА был идентифицирован в 1995 г., его обозначение SMN (survival motor neuron),
- В среднем один из 6000 детей рождается со SMA, в разных странах частота сильно различаются,
- 50 % детей с СМА не доживают до двух лет (это дети преимущественно с 1-й формой заболевания),
- SMA может проявиться в любом возрасте, «мягкие» формы проявляются в среднем и пожилом возрасте,
- Один из каждых 40 людей имеет рецессивный ген, способный вызывать SMA,
- В соответствии с менделевским раcщеплением ребёнок двух носителей поражается СМА с вероятностью 25 %. В этом случае оба родителя несут одиночный дефектный ген, но защищены присутствием второго, нормального гена, который является вообще достаточным для нормальной функции организма. Две дефектных копии гена приводят к генному нарушению, так как не обеспечивается синтез необходимого белка.
- В ходе медико-генетического обследования нескольких российских и среднеазиатских популяций (1,8 млн человек) выявлено 33 больных спинальной мышечной атрофией (СМА): 29 с детской проксимальной СМА (СМА I—III) и 4 c редкими формами. Выявлено «перекрывание» проявлений разных типов СМА I—III (I—II и II—III) у части больных, внутрисемейные различия типов в 3-х из 6-ти семейных случаев, клинико-генетический полиморфизм редких форм СМА. (Г. Е. Руденская, Р. А. Мамедова)
История и патогенез
Спинальная мышечная атрофия у детей впервые была описана G. Werdnig в 1891 году. G. Werdnig представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. J. Hoffmann обосновал нозологическую самостоятельность заболевания. В дальнейшем G. Werdnig и J. Hoffmann (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Е. Kugelberg и L. Welander выделили новую нозологическую форму спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной G. Werdnig и J. Hoffmann.
SMA вызвана мутацией в части ДНК, называемой SMN1 ген, который обычно производит белок SMN. Из-за мутации гена, у людей со SMA производится меньшее количество SMN белка, что приводит к потере моторных нейронов.
Классификация типов СМА
- Тип 1, или болезнь Werdnig-Hoffmann, — наиболее неблагоприятная форма SMA.
Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием. Тип 1 SMA проявляется у детей в период от рождения до 6 месяцев.
- Тип 2 — несколько более благоприятен.
Пациенты способны сидеть без поддержки или даже стоять с поддержкой и обычно не страдают при приёме пищи. Однако они имеют увеличенный риск осложнений от инфекций дыхательных путей. Тип 2 SMA проявляется у детей в период 7-18 месяцев.
- Тип 3, также известный как болезнь Kugelberg-Welander — наименее смертельная форма SMA детского возраста.
Пациент способен стоять, но испытывает сильную слабость, с тенденцией к инвалидизации (передвижение в коляске). Тип 3 SMA проявляется после 18 месяцев, реже — во взрослом состоянии.
- Тип 4 — взрослая форма болезни, признаки проявляются после 35 лет.
Признаки атрофии обычно начинаются в мышцах рук, ног и языка, затем распространяются к другим областям тела.
- Взрослое X-связываемое начало SMA, также известное как Синдром Кеннеди, или бульбарная спинномозговая мышечная атрофия, развивается только у взрослых людей. При этом заболевании заметно затронуты лицевые мышцы и мышцы языка. Кроме того, часто встречается гинекомастия. Прогноз болезни неопределённый, но обычно заболевание прогрессирует медленно.
Статистика форм SMA (2006)
Статистика приведена из Patient Registry Report, составитель Connie Garland для SMA Registry, The International Coordinating Committee for clinical trials in SMA:
- Количество семей: 1,386
- Число заболевших: 1,535
- Из них женщин — 759
- Из них мужчин — 776
- Умерло — 242
- Живы — 1,293
- Тип SMA
- Тип 1 — 489
- Тип 2 — 511
- Тип 3 — 315
- Adult Onset — 37
- Болезнь Кеннеди (Kennedy Disease) — 7
- Неясные / неустановленные формы — 176
Лечение
До последнего времени не было методов специфической терапии СМА.
Так как спинальная мышечная атрофия — нарушение, которое проявляется в синапсах моторных нейронов, состояние может быть улучшено за счёт увеличения уровня SMN — белка. Цель современных исследований — поиск препаратов, увеличивающих уровни SMN. Основные результаты получены пока в исследовательских группах США, Германии, Италии.
Предложено несколько препаратов (вальпроевая кислота, бутират натрия и др.), проводятся их клинические исследования в группах добровольцев. Сведений о результативном применении стволовых клеток пока нет.
Есть сведения, что физиопроцедуры, массаж и др. методы нейро-мышечной стимуляции (напр., дельфинотерапия) положительно сказываются на состоянии больных. В то же время сведений об отрицательных эффектах от физических нагрузок нет.
Больные СМА нуждаются в специальном диетическом питании, поддерживающей терапии и многих других попечительских действиях. Количество вопросов растёт, как снежный ком — только опытный врач может помочь сориентироваться во всех проблемах.
Для объединения больных со спинальной мышечной атрофией (СМА) на Украине инициировано открытие Благотворительного Фонда родителей детей со спинальной атрофией (см. ссылки внизу статьи). Регулярно проводятся телеконференции специалистов и родственников заболевших.
Профилактика
В настоящий момент возможна только пассивная профилактика — предупреждение родителей с риском СМА о возможных последствиях, пренатальный скрининг — диагностика формы СМА для принятия решения о рождении. Диагностика производится в некоторых медико-генетических центрах.
Внешние ссылки. Организации для помощи больным СМА
См. также
в том числе распространенные заболевания, проявления которых которые могут быть скорректированы путем приема комплекса специальных лекарственных препаратов, диетой, БАД
| Заболевания ЦНС | |
|---|---|
| Головной мозг Энцефалопатия | |
| Головная боль | Мигрень • Кластерные головные боли • Сосудистая головная боль • Головная боль напряжения |
| Расстройства сна | Бессонница • Гиперсомния • Апноэ во сне • Нарколепсия • Катаплексия • Синдром Клейне — Левина • Нарушения цикличности сна и бодрствования |
| Двигательные и экстрапирамидные расстройства | Дискинезия: Дистония • Хорея • Миоклония • Болезнь Унферрихта — Лундборга • Тремор (Эссенциальный тремор, Интенционный тремор) • Синдром беспокойных ног • Синдром мышечной скованности |
| Эпилептические припадки Эпилепсия | Локализованная эпилепсия • Генерализованная эпилепсия • Эпилептический статус • Миоклоническая эпилепсия • Туберозный склероз |
| Деменция | Болезнь Альцгеймера • Лобно-височная деменция/Лобно-височная лобарная дегенерация • Мультиинфарктная деменция |
| Цереброваскулярные болезни | Преходящие нарушения мозгового кровообращения (Гипертензивный церебральный криз, Транзиторная ишемическая атака) • Дисциркуляторная энцефалопатия (Церебральный атеросклероз, Подкорковая атеросклеротическая энцефалопатия, Хроническая гипертоническая энцефалопатия) • Инсульт (Ишемический инсульт, Внутримозговое кровоизлияние, Субарахноидальное кровоизлияние) • Тромбоз синусов твёрдой мозговой оболочки (Тромбоз кавернозного синуса) |
| Воспалительные заболевания | Абсцесс головного мозга • Менингит • Арахноидит • Энцефалит • Менингоэнцефалит • Энцефалит Расмуссена • Клещевой энцефалит |
| Демиелинизирующие заболевания | Аутоиммунные заболевания (Рассеянный склероз, Оптикомиелит, Болезнь Шильдера) • Наследственные заболевания (Адренолейкодистрофия, Болезнь Краббе) • Центральный понтинный миелинолиз • Синдром Маркиафавы — Биньями • Синдром Альперса |
| Системная атрофия | Болезнь Пика • Болезнь Хантингтона • Спинномозговая атаксия Спинальная мышечная атрофия: Синдром Кеннеди • Спинальная мышечная атрофия у детей • Болезнь двигательного нейрона • Синдром Фацио-Лонде • Боковой амиотрофический склероз |
| Митохондриальные заболевания | Синдром Лея |
| Опухоли | Опухоль головного мозга • Туберозный склероз |
| Спинномозговая жидкость | Внутричерепная гипертензия • Отёк мозга • Внутричерепная гипотензия |
| Травмы | Черепно-мозговая травма (Сотрясение мозга, Ушиб головного мозга, Диффузное аксональное повреждение головного мозга) |
| Другие заболевания | Спинномозговая грыжа • Синдром Рея • Печёночная кома • Токсическая энцефалопатия • Гематомиелия |
| Спинной мозг Миелопатия | |
| Воспалительные заболевания | Менингит • Арахноидит • Менингоэнцефалит • Миелит • Полиомиелит • Демиелинизирующие заболевания • Тропический спастический парапарез |
| Другие заболевания | Сирингомиелия • Сирингобульбия • Синдром Морвана • Сосудитая миелопатия • Спинальный инсульт • Сдавление спинного мозга • Энцефаломиелит |
dic.academic.ru